Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/004
REVIEW ARTICLE OPEN ACCESS
Salvinorin A: A Mini Review of Physical and Chemical Properties Affecting Its Translation from Research to Clinical Applications in Humans
Edward Orton1 , PhD, Renyu Liu2, MD, PhD
Edward Orton, Ph.D. Chief Scientific Officer, Cetazam Therapeutics. 3160 Chestnut Street, Suite 200 Philadelphia PA 19104, E-mail: edward.orton@gmail.com
Received: May 01, 2014 | Accepted: June 15, 2014 | Published: July 01, 2014
Citation: Orton E, Liu R. Salvinorin A: A Mini Review of Physical and Chemical Properties Affecting Its Translation from Research to Clinical Applications in Humans1Cetazam. Trans Periop & Pain Med 2014, 1(1):9-11.
Salvinorin A is a potent and selective agonist of kappa opioid receptors in the brain. Recent studies in several animal models have revealed that Salvinorin A has anti-addiction, anti-depression properties and exhibits pronounced neuroprotective effects against hypoxia/ischemia induced brain damage, and have raised interest in potential clinical applications in several acute pathologies involving oxygen deficiency in the brain. This review focuses on the chemical and physical properties of Salvinorin A and their impact on development of a rational formulation to enable its translation from a research compound to a novel therapeutic agent.
Introduction
Salvinorin A (SA) is a psychoactive neoclerodane diterpenoid (Figure 1a) isolated from the plant Salvia divinorin, a member of the Sage family that occurs naturally in Mexico[1]. As a recreational drug, SA is typically either smoked or the leaves are chewed to allow absorption of the active drug through the mucous membranes of the oral cavity and giving rise to short lived intense hallucinations, antinociception, sedation and dysphoria. A highly potent drug, Salvinorin A shows central nervous system (CNS) activity at doses of 200-500 µg [1-3]. After PO dosing in humans, SA is rapidly degraded by enzymes in the intestinal tract resulting in complete loss of exposure [1]. Similarly, the efficacy of SA as a hallucinogen via buccal or sub-lingual routes is diminished in saliva. Salvinorin A has also been shown to be a substrate for P-glycoprotein efflux transporters that are highly expressed in the blood brain barrier, and this contributes to the short cerebral residence time of SA[4].
Since the discovery of SA as the only known naturally-occurring, non-nitrogenous kappa opioid receptor (KOR) agonist (Figure 1b) [5], research leading to clinical use of SA as a potential novel medication for addiction [6,7], depression management [8], and as a neuroprotective agent for neonatal hypoxia/ischemia, as well as brain injury due to oxygen insufficiency arising from cardiac arrest and stroke has been described in recent literature [9]. Specific cerebrovasodilation effects were observed in a newborn pig model which indicated that Salvinorin A is a fastonset, short-lived agent that may find therapeutic applications in a number of acute brain pathologies accompanied by vasoconstrictive events [10].
Figure 1: a) Molecular structure of Salvinorin A. b) Salvinorin A occupies the binding site in the kappa opioid receptor. The binding site prediction was carried out using DockingServer20 as previously described [21]. The coordinates of the crystal structure of the kappa receptor were obtained from the protein data bank (PDB) with access code of 4DJH [22]. The image was generated using PyMOL (Version 1.5.0.4, Schrodinger LLC, New York, NY).
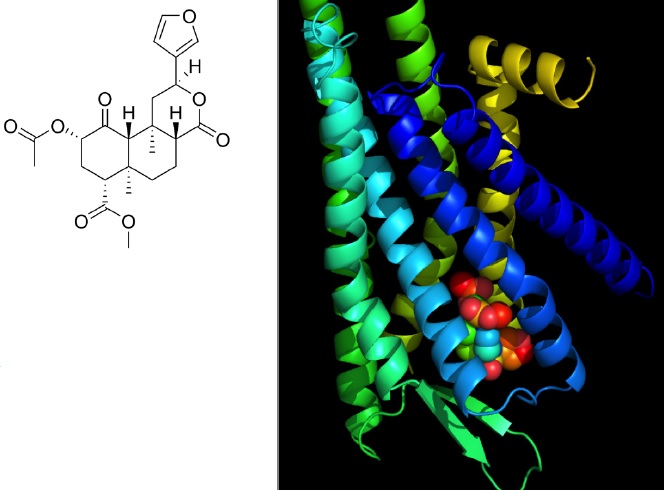
In this review we discuss the chemical and physical properties of Salvinorin A and their roles in the process of defining a rational translation pathway for formulating the drug substance into a clinically deliverable dosage form for acute medical situations.
Chemical and Physical Properties
Physical and chemical properties of SA (see Table 1) reveal a compound with subtle and potentially complex issues from a formulation perspective.
Table 1: Physical properties of Salvinorin A.
| Property | Value | Comments |
|---|---|---|
| Molecular weight | 432.464 g/mol | Anhydrous |
| Molecular formula | C23H28O8 | Anhydrous |
| Crystal hydrate molecular formula | C23H28O8 .1/3 H2O | Trienhydrate crystal form |
| Melting point | 238-240 ℃ | Trienhydrate crystal form |
| Calculated LogP | 2.49 | ChemAxon/Marvin |
| pKa | None | Non-ionizable |
| Chiral centers | 7 | 3 epimerizable |
| Optical rotation | -41 deg C at 25 ℃ | c = 1 in CHCl3 |
| Water content | 1.4% (Karl Fischer) | Trienhydrate crystal form |
Lacking ionizable functional groups, Salvinorin A cannot form soluble salts. SA has eight hydrogen bond acceptor sites, all oxygen atoms, and no hydrogen-bond donor groups. It would thus appear likely that its crystal lattice should comprise only weak non-bonded interactions between adjacent molecules and a resultant diminished lattice energy reflected in a low melting point [11]. In fact, SA has a rather high melting point range reported as 238-240 ℃. An x-ray crystal structure of SA revealed that crystallization from aqueous organic solvents (acetone, methanol) yields SA as a stoichiometric hydrate with one molecule of water per three molecules of Salvinorin A [12]. Unfortunately, the x-ray structure did not resolve the positions of the water hydrogen atoms, so a detailed picture of the hydrogen-bonding linking SA molecules into the crystal lattice remains unclear. The existence of Salvinorin A as a high melting crystalline hydrate can be expected to influence its solubility behavior in organic and aqueous solvents Poor solubility will likely limit formulation approaches accessible for translation of the drug into a clinically applicable medicine.
The chemical stability of Salvinorin A has not been examined in published literature to date. Nonetheless, any clinically relevant formulation will need to circumvent hydrolytic conditions that may affect the ester and lactone moieties essential to the activity of SA at the KOR receptors. For example, hydrolysis of the acetate in SA produces the well-known derivative Salvinorin B which lacks KOR affinity [13]. Further, the presence of three epimerizable carbons in SA raises concerns over the stereochemical integrity of the drug substance. Chromatographic and spectroscopic analytical methods adequate to detect epimers of SA have been reported [14]. In addition, direct photochemical degradation of SA is not a concern since its ultraviolet absorption onset (due to the furan chromophore) is less than 220 nm.
Rational Formulation Approaches for Salvinorin A
In view of Salvinorin A's susceptibility to enzymatic degradation as well as being a substrate for P-glycoprotein efflux, the PO route of administration is clearly not viable. Not surprisingly due to the poor aqueous solubility of Salvinorin A, intravenous (IV) formulations described in numerous in vivo studies over the past 15 years have largely employed pharmaceutically unacceptable solvents (dimethylsulfoxide, methanol and acetone). IV formulations using moderately high percentages of ethanol and propylene glycol [14] as co-solvent may be acceptable for acute clinical situations [15]. The IV route permits some control over duration of the therapeutic dosing level, a matter of concern for the psycho active side effects of Salvinorin A. An IV nanocrystal formulation of SA may be an alternative to the co-solvent approach, especially when protracted therapeutic brain levels of the neuroprotectant drug are desired.
Despite the poor apparent bioavailability of Salvinorin A when dosed via the oral cavity by recreational drug users [4], for acute dosing this remains a potential route of administration. Ready access to the oral cavity is a clear advantage for buccal delivery, as is the ease of administration even in the absence of medical personnel. In principle, high doses of SA could be delivered through the oral mucosa using mucoadhesive gels or patches in combination with penetration enhancers to achieve therapeutic levels of SA in the brain. Other advances in buccal delivery systems have been described recently [16]. Suspensions of Salvinorin A as micronized or nanocrystalline particles would be compatible with such delivery systems and act, in effect, to provide prolonged exposure of SA to the brain.
Intranasal administration for direct to brain drug delivery offers several important advantages over other administration routes: Rapid onset of therapeutic activity; bypassing the blood brain barrier; avoiding hepatic first pass metabolism [17]. In the case of Salvinorin A for acute neuroprotective therapy a multitude of dosing devices are currently in use (or in development) that can enable initiation of therapy by non-medical persons. The existence of a plurality of intranasal delivery systems that can deliver SA suspension formulations make this an attractive approach. A clear shortcoming of intranasal delivery may be anticipated in patient to patient variability of response due to a combination of intrinsic (e.g. anatomical features) and extrinsic (e.g. accuracy of dosage delivery to site) factors.
Inhalation of Salvinorin A via smoking or vaporization is well known to provide very rapid brain uptake and intense psychotropic effects [18,19]. Inhalation of medicines is a well-developed field and a large variety of delivery devices and formulations are currently marketed. However, in the context of acute treatment of the unconscious and/or unresponsive patient, inhaled dosage delivery appears problematic at least outside the hospital setting.
Conclusions
In this review we have examined the chemical and physical properties of Salvinorin A, a potential novel drug for neurological diseases, to define viable approaches to developing formulations that can be translated to clinically relevant dosage forms for treatment of acute onset illnesses outside a hospital setting. Salvinorin A exists as a stable stoichiometric hydrate and exhibits very poor solubility in aqueous vehicles. Intravenous administration may be achievable with elevated co-solvent vehicles or nanocrystalline suspensions, although its utility in the absence of medical staff is doubtful. Dosage forms targeting the oral mucosa appear worthy of further investigation: while the bioavailability is low, the ability to readily deliver high doses to the oral cavity (a highly accessible site) may allow maintenance of therapeutic drug levels in the brain. Intranasal drug delivery affords rapid, direct access of the drug to the brain bypassing the hepatic circulation. A multitude of choices for formulation and delivery devices exist, but patient to patient variability to this route of drug delivery remains uncertain particularly for administration by non-medical persons. Preferred among recreational drug users, delivery of Salvinorin A via inhalation offers clear advantages in terms of rapidly reaching therapeutic drug levels in the brain. For the purposes of acute treatment of an unresponsive patient, inhalation delivery may not be suitable.
Disclosure of Funding
This work is supported by departmental funding from the department of Anesthesiology and Critical Care at University of Pennsylvania and support from NIH K08-GM-093115(PI:RL).
References
- Siebert DJ: Salvia divinorum and salvinorin A: new pharmacologic findings. Journal of ethnopharmacology 1994; 43: 53-6.
- Johnson MW, MacLean KA, Reissig CJ, Prisinzano TE, Griffiths RR: Human psychopharmacology and dose-effects of salvinorin A, a kappa opioid agonist hallucinogen present in the plant Salvia divinorum. Drug and alcohol dependence 2011; 115: 150-5.
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, D'Souza DC: Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the kappa opioid agonist Salvinorin A in humans. Biological psychiatry 2012; 72: 871-9.
- Butelman ER, Caspers M, Lovell KM, Kreek MJ, Prisinzano TE: Behavioral effects and central nervous system levels of the broadly available kappa agonist hallucinogen salvinorin A are affected by P-glycoprotein modulation in vivo. The Journal of pharmacology and experimental therapeutics 2012; 341: 802-8.
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB: Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proceedings of the National Academy of Sciences of the United States of America 2002; 99: 11934-9.
- Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, Dersch CM, Rothman RB, Kivell B, Prisinzano TE: Potential Drug Abuse Therapeutics Derived from the Hallucinogenic Natural Product Salvinorin A. MedChemComm 2011; 2: 1217-1222.
- Morani AS, Kivell B, Prisinzano TE, Schenk S: Effect of kappa-opioid receptor agonists U69593, U50488H, spiradoline and salvinorin A on cocaine-induced drug-seeking in rats. Pharmacology, biochemistry, and behavior 2009; 94: 244-9.
- Harden MT, Smith SE, Niehoff JA, McCurdy CR, Taylor GT: Antidepressive effects of the kappa-opioid receptor agonist salvinorin A in a rat model of anhedonia. Behavioural pharmacology 2012; 23: 710-5.
- Wang Z, Ma N, Riley J, Armstead WM, Liu R: Salvinorin A administration after global cerebral hypoxia/ischemia preserves cerebrovascular autoregulation via kappa opioid receptor in piglets. PloS one 2012; 7: e41724.
- Su D, Riley J, Kiessling WJ, Armstead WM, Liu R: Salvinorin A produces cerebrovasodilation through activation of nitric oxide synthase, kappa receptor, and adenosine triphosphate-sensitive potassium channel. Anesthesiology 2011; 114: 374-9.
- Infantes L, Fabian L, Motherwell WDS: Organic crystal hydrates: what are the important factors for formation. Cryst Eng Comm 2007; 9: 65-71.
- Ortega A, Blount JF, Manchand PS: Salvinorin, a new trans-Neoclerodane diterpene from aalvia divinorurn (Labiatae). J Chem Soc Perkin Trans I 1982: 2505-2508.
- Prisinzano TE: Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life sciences 2005; 78: 527-31.
- Giner JL, Kiemle DJ, Kutrzeba L, Zjawiony J: Unambiguous NMR spectral assignments of salvinorin A. Magnetic resonance in chemistry : MRC 2007; 45: 351-4.
- Demey HE, Daelemans RA, Verpooten GA, De Broe ME, Van Campenhout CM, Lakiere FV, Schepens PJ, Bossaert LL: Propylene glycol-induced side effects during intravenous nitroglycerin therapy. Intensive care medicine 1988; 14: 221-6.
- Chinna Reddy P, Chaitanya KS, Madhusudan Rao Y: A review on bioadhesive buccal drug delivery systems: current status of formulation and evaluation methods. Daru : journal of Faculty of Pharmacy, Tehran University of Medical Sciences 2011; 19: 385-403.
- Dhuria SV, Hanson LR, Frey WH, 2nd: Intranasal delivery to the central nervous system: mechanisms and experimental considerations. Journal of pharmaceutical sciences 2010; 99: 1654-73.
- Addy PH: Acute and post-acute behavioral and psychological effects of salvinorin A in humans. Psychopharmacology 2012; 220: 195-204.
- MacLean KA, Johnson MW, Reissig CJ, Prisinzano TE, Griffiths RR: Dose-related effects of salvinorin A in humans: dissociative, hallucinogenic, and memory effects. Psychopharmacology 2013; 226: 381-92.
- Bikadi Z, Hazai E: Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform 2009; 1: 15.
- Liu R, Perez-Aguilar JM, Liang D, Saven JG: Binding site and affinity prediction of general anesthetics to protein targets using docking. Anesth Analg 2012; 114: 947-55.
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC: Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012; 485: 327-32.