Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/024
REVIEW ARTICLE OPEN ACCESS
Updated Mechanisms of Sickle Cell Disease-Associated Chronic pain
Brianna Lutz1, B.S., Steffen E. Meiler2, M.D., Ph.D., Alex Bekker1, MD, PhD, Yuan-Xiang Tao1,3 , MD, PhD
1Department of Anesthesiology, Rutgers Graduate School of Biomedical Sciences, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ, USA
2Department of Anesthesiology and Perioperative Medicine, Georgia Regents University, Augusta, GA, USA
3Departments of Cell Biology & Molecular Medicine, Pharmacology & Physiology, and Neurology & Neuroscience, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ ,USA
Yuan-Xiang Tao, MD, PhD Department of Anesthesiology Rutgers New Jersey Medical School, Rutgers, The State University of New Jersey 185 S. Orange Ave., MSB, F-548, Newark, NJ 07103. Tel: +1-973-972-9812; Fax: +1-973-972-1644, E-mail: yuanxiang.tao@njms.rutgers.edu
Editor: Renyu Liu, MD; PhD Associate Professor Director of Preoperative Medicine Department of Anesthesiology and Critical Care Perelman School of Medicine at the University of Pennsylvania 336 John Morgan building, 3620 Hamilton Walk, Philadelphia, PA 19104, Email: liur@uphs.upenn.edu
Received: June 15, 2015 | Accepted: July 17, 2015 | Published: July 26, 2015
Citation: Lutz B, Meiler SE, Bekker A, Tao YX. Updated Mechanisms of Sickle Cell Disease-Associated Chronic pain. Transl Perioper & Pain Med 2015; 2(2): 8-17.
Abstract
Sickle cell disease (SCD), a hemoglobinopathy, can cause sickling of red blood cells, resulting in vessel blockage, stroke, anemia, inflammation, and extreme pain. A vast majority of SCD patients experience pain on a chronic basis, and many turn to opioids to provide limited relief. The side effects that come with chronic opioid use push for research into understanding the specific mechanisms of SCD-associated chronic pain. Current advances in SCD-associated pain have focused on alterations in the pain pathway including nociceptor sensitization and endogenous pain inducers. This article reviews the underlying pathophysiology of SCD, potential pain mechanisms, current treatments and their mechanism of action, and future directions of SCDassociated pain management. The information provided could help propel research in SCD-associated chronic pain and uncover novel treatment options for clinicians.
Introduction
Sickle cell disease (SCD) affects around 100,000 Americans and accounts for over $450 million in healthcare expenses each year [1,2]. SCD is a group of disorders associated with a mutation in the β globin gene [1,2]. In red blood cells, two β globins combine with two α globins to form the oxygen carrying molecule hemoglobin. Upon deoxygenation, sickle β globin polymerizes, which causes the blood cell to take on a half-moon shape, adhere to blood vessels, and hinder blood flow. These vaso-occlusive events lead to organ damage, extreme pain, stroke, and a shortened lifespan. Sickled red blood cells also die faster than normal red blood cells, resulting in hemolytic anemia [3]. Excellent reviews on the pathophysiology and genetics of SCD have been previously published [4-6]. This review focuses on the possible underlying mechanisms of SCD-associated chronic pain, clinical treatment options, novel therapies and their proposed mechanisms, and future directions. A literature review search was conducted using the PubMed database with keywords “sickle cell disease and chronic pain”. Selection criteria were based on the impact of the study and the year in which it was published. The goal of this review is to summarize the most recent advances in the realm of Sickle Cell Disease-associated chronic pain. Since pain in SCD patients is associated with an increase in morbidity [7], understanding the cause of SCD-associated pain may provide insight into SCD itself.
Clinical characterization of SCD-associated pain
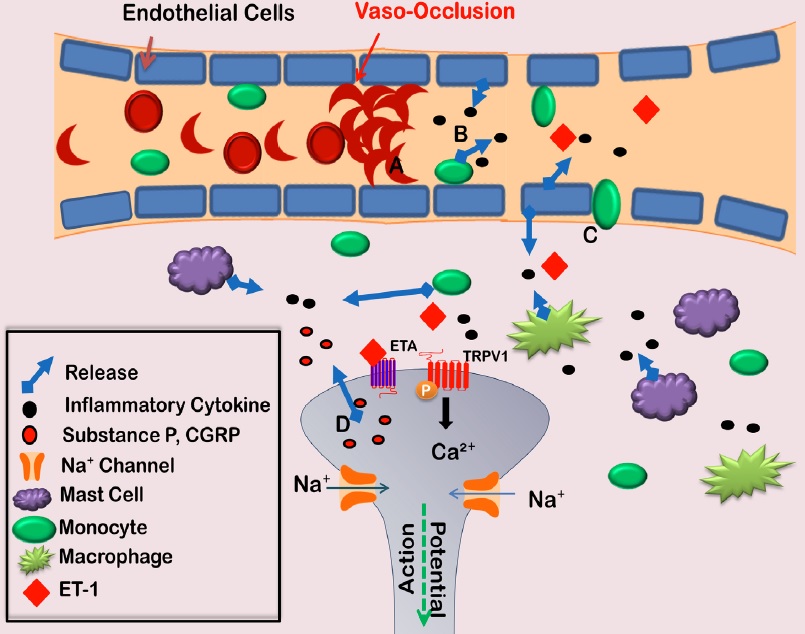
Acute painful episodes are the main cause of SCD related hospitalizations [8,9]. The cause of these episodes may be vaso-occlusive (VOC) events, in which sickled cells adhere to blood vessels and block blood flow [10]. The interaction between sickled red blood cells and the vessel wall causes endothelial cell activation, which leads to the production of inflammatory mediators. These mediators are thought to play a role in SCD pathology (Figure 1) [11,12]. Extravasation also occurs as endothelial cell permeability increases. The increase in monocytes, mast cells, and macrophages in the surrounding tissue contributes to the inflammatory milieu triggering nociceptor activation. The consequence of these events is an acute painful episode, which is often severe enough to require an emergency room visit and intravenous opioids [13].
Figure 1: Vaso-occlusive events lead to acute painful episodes in SCD patients. Deoxygenation of red blood cells harboring sickle beta globin causes polymerization of hemoglobin and morphological changes in the red blood cell. The cell adheres with greater affinity to endothelial cells lining blood vessel walls, creating a blockage (A). Endothelial cells then become activated releasing cytokines (B) and allowing for increased extravasation of monocytes (C). The increase of inflammatory cells in the surrounding tissue further contributes to the release of cytokines surrounding nociceptor terminals. This inflammatory soup then activates the nociceptor to allow the release of substance P or CGRP from nociceptor terminals, resulting in a feed-forward mechanism contributing to nociceptor sensitization (D). CGRP: calcitonin gene related peptide; TRPV1: transient receptor potential vanilloid channel 1.
In addition to acute painful episodes that are prevalent in SCD patients, chronic pain is often reported. In a recent survey of 232 SCD patients, pain was self-reported on 56% of 31,017 total diary days [14]. Painful crisis (VOCs) was reported on 12.7% of these days [14]. In a separate survey of 227 SCD patients, 92% reported pain that lasted from 6 months to more than 2 years [15]. Chronic pain in patients may stem from a known cause including edema, ischemia, or nerve damage; however, intractable chronic pain exists in SCD patients as well. Understanding the underlying causes of intractable chronic pain is of interest for the purpose of developing novel therapies.
In clinical studies, SCD patients with chronic pain exhibit symptoms that suggest sensitization of pain pathways. Painful episodes increase during times of cold weather and windy weather, suggesting a possible cool/cold sensitivity in SCD patients [16,17]. In clinical tests, patients with SCD-associated chronic pain show increased sensitivity to cold and heat compared to the matched control patients [18,19]. Brandow et al. reported that the median heat painful threshold for SCD patients was 42.7 ℃ compared to 45.2 ℃ in the matched controls. The cool/cold threshold for SCD patients was 21.1 ℃ compared to 14.8 ℃ for the matched controls. These significant differences demonstrate thermal hyperalgesia and cool/cold allodynia in SCD patients.
Current clinical treatment of SCD-associated pain
Current treatments for SCD-associated chronic pain do not differ from the common treatment strategies for other chronic pain conditions, in which opioids are often the most prescribed therapeutic option [20,21]. In a survey of 299 SCD patients, opioid use was reported on almost 60% of a total of 16,818 diary days [22]. However, opioids produce severe side effects, including hyperalgesia and tolerance [23,24]. Opioids may even produce severe pathological changes in SCD patients that could contribute to organ damage [25]. More targeted treatment options could reduce the need for opioids, and their harmful side effects.
Although opioids are the common treatment for SCD-associated pain, other therapeutics are also being considered. Hydroxyurea therapy is recommended for SCD patients with recurring vaso-oclussive episodes [26]. Hydroxyurea is a ribonucleotide reductase inhibitor that raises the level of fetal hemoglobin in SCD patients [26]. Daily oral therapy of hydroxyurea has been demonstrated to reduce SCD associated complications including pain [26,27] Hydroxyurea is associated with several side effects including neurological, gastrointestinal, and cutaneous side effects [28]. Additionally, hydroxyurea therapy is not a targeted treatment for SCD associated chronic pain, thus more efficient treatment options should be considered. Histone deacetylase inhibitors also elevate fetal hemoglobin levels, but their clinical efficacy has yet to be validated in SCD patients [29].
Recently, a calcium/calmodulin protein kinase IIα (CaMKIIα) inhibitor was used in a clinical trial of its adverse effects and pain relief specific to the SCD patients. Fifteen out of a total of 18 SCD subjects reported a 50% reduction in pain intensity as determined by a Visual Analogue Scale for pain intensity [30]. It was reported that CaMKIIα inhibitors may reduce pain sensitivity by preventing the phosphorylation of N-metyl-D-aspartate (NMDA) receptors and subsequent activation and calcium influx [31]. TRPV1 may be a downstream target of this inhibitor as CaMKIIα can activate TRPV1 channels [31]. CaMKIIα inhibitors produced few side effects in SCD patients, however, painrelief was not complete, and many patients still reported chronic pain. Adjunct therapies or other targeted treatments are still required.
Similar to chronic pain patients, SCD patients are looking to alternative therapeutics such as acupuncture for pain management [15]. In a recent survey of 24 SCD patients who receive acupuncture therapy for a painful crisis, 9 patients who received inpatient acupuncture at a single institution reported pain reduction immediately following one session of acupuncture (pain score reduction of 2.1 points) [32]. There are some limitations to this study, however, including a small sample size, and further research must be done to show that acupuncture is a preferable treatment option for SCD-associated pain.
As an adjunct therapy, cognitive behavioral therapy (CBT) is gaining popularity in pediatric sickle cell disease. A recent study conducted by Schatz, et al. analyzed the effect of CBT coping skills training on pain perception in SCD youth [33]. 46 SCD patients received one session of CBT followed by 8 weeks of smartphone administered CBT training. Participants completed daily diaries and questionnaires which were used to access pain intensity and functional activity. The daily diaries revealed that CBT skills reduced next-day pain intensity suggesting that the use of coping strategies may reduce pain intensity in SCD patients. CBT may be a beneficial adjunct therapy for SCD chronic pain, but further studies do need to be completed to address the effect of CBT in adult patients.
Exercise may also be a beneficial adjunct therapy for SCD associated pain; however, exercise in SCD patients is controversial. One case report by Tinti, et al. found a reduction in pain in a 32 year old SCD female following an aquatic rehabilitation program [34]. This mild exercise program incorporated stretching, aerobic exercise, and relaxation administered in two 45 minute sessions per week for 5 weeks. In a separate clinical study, oxidative stress markers in SCD patients following mild to moderate exercise did not differ from healthy controls [35]. This study was careful to acknowledge, however, that some SCD patients displayed increases in endothelial activation and oxidative stress which could lead to vaso-occlusive events. Exercise in SCD patients can produce severe complications including pulmonary hypertension and vaso-occlusive events [36-38], therefore, exercise as a treatment for SCD associated pain is approached with great caution [39]. Further research incorporating larger sample sizes needs to be conducted.
Taken together, it appears that current treatments for SCD-associated pain are limited and/or ineffective. Further research on the mechanisms that underlie this disorder is required and may provide a new strategy for its therapy.
Animal models of SCD-associated pain
Research into the complex pathophysiology of SCDassociated pain has taken great strides due to the development of animal models [40]. Two common mouse models of SCD are the Berkeley [41] and Townes [42] models. In both of these models, the mouse α and β-globin alleles have been knocked down, and human α and human sickle β-globin alleles have been inserted. Thus, these mouse models express only human α globin and sickle β-globin, mimicking human SCD genotypes. The Townes model differs from the Berkeley model in that the transition from fetal hemoglobin (γ-globin) expression to sickle β-globin expression mimics the timeline found in humans. In Berkeley mice, γ-globin is replaced by sickle β-globin by 15 days gestation. This leads to increased perinatal lethality translating into small litter sizes, which are a major limitation of this model. In Townes mice, the transition is complete at 1 month of age, allowing for increased perinatal survival. Control SCD mice (HBAA) are also available, in which only human α and normal human β globin alleles are expressed [40,42].
The phenotypes in SCD mice are highly similar to those observed in SCD patients. Both Berkley (HBSSBERK) and Townes (HBSS) SCD mice display many of the pathophysiological changes observed in SCD patients including anemia, organ damage, hypoxia-induced cell sickling, and pain sensitivity [42,43,45,46]. Exposure to a hypoxic environment induces cell sickling in SCD mice, and can be used in the laboratory to induce a vaso-occlusive episode [40]. A major limitation of both SCD models is the severity of the disease. In SCD patients, the severity of the disease depends on a multitude of factors including γ-globin expression, β-globin expression (both normal and sickle), α-globin expression, and environment. SCD mice, on the other hand, are designed to express sickle β-globin and human α-globin exclusively, which translates into severe SCD. For this reason, translational issues may arise making it difficult to find effective therapeutics.
Behavioral studies using both SCD mouse models have revealed pain hypersensitivity similar to that observed in SCD patients. HBSS-BERK mice display increased heat and cold hyperalgesia in addition to mechanical allodynia [47]. Mechanical testing using von Frey filaments displayed a reduced mechanical threshold in HBSS-BERK mice when compared to HBAA mice. Thermal paw withdrawal latencies recorded by applying a heat source to the hindpaw of HBSSBERK and HBAA mice showed that HBSS-BERK mice responded to the heat stimuli faster than HBAA mice. The same decreased paw withdrawal latencies were observed in the HBSS-BERK mice when exposed to a cold surface (0 ℃) [47]. Cold hypersensitivity was also reported in Townes mice similar to that observed in HBSS-BERK mice [49]. Research thus far shows no differences in pain behavior between HBSS-BERK and Townes mice. Pain hypersensitivity also increases with age in SCD mice [50], which correlates with the phenomena observed in SCD patients [19].
Potential mechanisms of SCD-associated pain
Increased nociceptor activation, a characteristic of sensitization [51], was found in SCD animal models. Electrophysiological recordings of primary sensory neurons from SCD mice show enhanced nociceptor activity in response to painful stimuli. In an ex-vivo saphenous nerve preparation from HBSS-BERK and control mice, Zappia et al found that HBSS-BERK C-fibers responded at warmer temperatures when compared to control fibers during a decreasing temperature ramp from 32 ℃ to 2 ℃ (19.2 ± 1.2 ℃ vs. 14.6 ± ℃) [50]. This could represent sensitization of peripheral afferent terminals to cold stimuli as the control fibers can withstand colder temperatures without firing, thereby explaining cold hypersensitivity behavior observed in SCD mice. In a separate study of HBSS-BERK mice, mechanical allodynia was found to result from enhanced activation of mechanoreceptors. In an ex-vivo preparation of saphenous nerve, mechanical stimulation of HBSS-BERK Aδ mechanoreceptors and unmyelinated C fiber nociceptors produced increased action potential firing when compared to HBAA mice. To analyze similarity in phenotype between HBSS-BERK mice and SCD patients, Garrison et al used light touch stimulation to measure mechanical allodynia and nerve fiber response. HBSS-BERK mice showed a 1.7 fold increase in response to light touch compared to HBAA mice [52]. This light touch resembles soft strokes or wind, which have been reported to cause pain in SCD patients [16,17]. Additionally, using sine-wave electrical stimulation, Kenyon et al, found that HBSS-BERK and HBSS mice possess reduced threshold firing in sensory fibers (Aδ, Aβ, and C fibers) [49]. The sensitization of both un-myelinated and myelinated sensory fibers could explain the thermal hypersensitivity and mechanical sensitivity displayed by SCD mouse models and patients [18,19]. These studies identified sensitization of sensory fibers in sickle cell mice.
Peripheral sensitization may not be the only contributor to SCD associated chronic pain. Central sensitization, the phenomenon in which excitability of spinal cord neurons increases, may occur in SCD mouse models. Cataldo et al. recently found an increase in spontaneous firing, receptor field size, and electrophysiological responses to low-threshold stimuli and mechanical stimuli in spinal dorsal horn neurons of HBSS-BERK SCD mice when compared to HBAA-BERK control mice [53]. The increases in several known signaling pathway components were observed including phosphorylated ERK, p38, TLR4, and IL-6. ERK phosphorylation has been shown to lead to decreased potassium currents associated with the Kv4.2 potassium channels translating into hyperexcitability of dorsal horn neurons [54]. The chronic nature of SCD associated pain may be attributed to a combination of both peripheral and central sensitizations.
The question remains as to what is causing enhanced peripheral and central sensitizations in SCD mice and patients. Vincent et al. reported that mast cell activation in the periphery of HBSS-BERK mice with hyperalgesia contributes to the release of inflammatory mediators such as tryptase, substance P (SP) and calcitonin-gene related peptide (CGRP) [55]. Tryptase activates protease activated receptor 2 (PAR2) on peripheral nociceptors [55]. The latter can lead to activation of the transient receptor potential vanilloid 1 cation channels (TRPV1) and thus increased action potential firing in addition to CGRP and SP release [56]. The feed-forward mechanism induced by SP and CGRP release sensitizes nociceptors, producing pain to normally non-painful stimuli [57]. Substance P has also been found to be upregulated in blood plasma of SCD mice with cold hypersensitivity, and in blood serum from SCD patients [58]. PCR analysis of the DRG from HBSS-BERK and HBAA mice revealed an upregulation of Tachykinin receptor 1 and endothelin 1(ET-1) [50]. Substance P, the primary ligand of Tachykinin receptor 1, is known to play a role in pain sensation and is linked to spinal cord injury and cutaneous neurogenic inflammation [59]. The upregulation of its receptor in HBSS mice may represent an increase in SP activity.
In addition, ET-1, a known pain inducer, was also reported to be involved in pain in both preclinical and clinical studies [60,61]. ET-1 is a 21-amino acid peptide produced by multiple cell types including macrophages [62], neurons, and endothelial cells [63,64]. It is released in response to endothelial cell activation, an event that occurs during vaso-occlusive episodes. Other factors that induce ET-1 production include proinflammatory cytokines, growth factors, angiotensin II, mechanical stress, peripheral tissue injury, and hypoxia [65,66]. Elevated levels of ET-1 in the blood plasma of SCD patients and mice both during and after a vaso-occlusive episode [66] suggest that this peptide may play a role in SCD associated pain.
How peripheral ET-1 participates in enhanced nociceptor sensitization in SCD mice and patients is elusive. Preclinical and clinical studies have found that ET-1 can induce pain [60,67-69]. When ET-1 was administered subcutaneously into the forearm of human subjects, patients developed cold and mechanical hyperalgesia [70]. In rats, hindpaw administration of ET-1 resulted in thermal hyperalgesia and tactile allodynia [71,72], which were abolished with pretreatment of an endothelin-type A (ETA) receptor antagonist. The painful effects of ET-1 administration are linked to its binding to the endothelintype A receptor found on nociceptors [73,74]. The interaction of ET-1 and ETA receptors can induce nociceptor activation in pre-clinical studies of normal subjects [71,75]. ET-1 subcutaneous administration was found to activate C-fibers and ATM-fibers in rat sciatic nerve in a dose-dependent manner [67] This activation was blocked when an ETA receptor antagonist was administered in conjunction with ET-1. ET-1 has also been shown to activate C-fibers in microneurography studies of human C-fibers [68]. C-fibers and ATM--fibers correspond with small peptidergic nociceptors, an area of high ETA receptor localization. Increased nociceptor activation correlates with behavioral responses as demonstrated in rats that were injected with ET-1 in the plantar tissue of hindpaws. Following ET-1 injection, increased hindpaw flinching, a behavioral display of nociception [67], occurred. Additionally, when the ETA receptor is stimulated, hyperpolarization of voltagedependent tetratoxin-resistant sodium channels occurs, which are exclusively located in small DRG neurons [76]. Since peripheral ET-1 is elevated in SCD patients, it could activate nociceptors and cause pain hypersensitivity as observed in the above ET-1 induced pain studies.
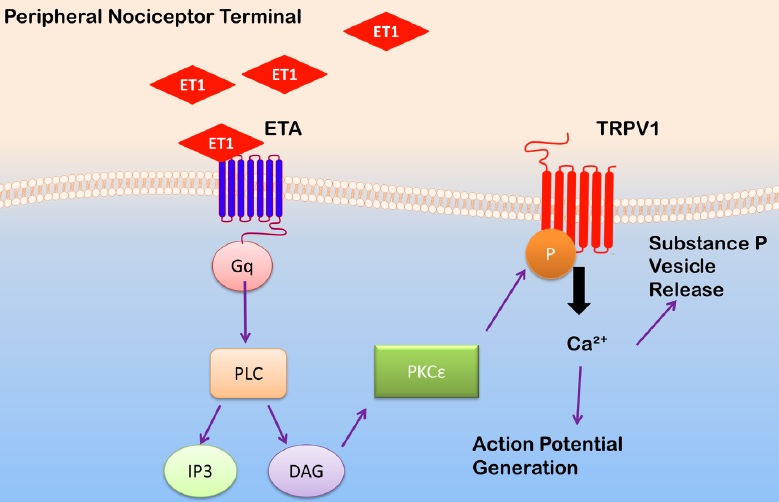
ET-1 may sensitize nociceptors by acting on transient receptor potential vanilloid 1 (TRPV1) channels, which co-localize with ETA receptors in peripheral nociceptors [72,77]. TRPV1 channels are non-selective cation channels with high affinity for calcium. Noxious temperatures (in vivo, greater than 43oC) and the compound capsaicin are known activators of TRPV1 channels [78]. Inflammatory mediators, such as those released following ischemia/reperfusion, can lower the activation threshold of TRPV1 channels [78]. The accumulation of inflammatory mediators, including ET1, in SCD may lead to sensitization of TRPV1 channels [79]. Indeed, in preclinical studies, subcutaneous injection of ET-1 sensitizes TRPV1 channels, leading to prolonged mechanical allodynia [72]. When TRPV1 antagonists are administered prior to ET-1 injection, mechanical allodynia is quickly attenuated. Additionally, administration of the TRPV1 antagonist capsazepine attenuated ET-1 induced thermal hyperalgesia in rats [71]. In electrophysiology studies, ET-1 was found to potentiate capsaicin-induced TRPV1 current in HEK293 cells expressing both ETA receptors and TRPV1 channels [80]. Based on these studies, ETA receptors may mediate ET-1 nociceptor activation by activating TRPV1 channels (Figure 2).
Figure 2: Proposed mechanism that underlies ET-1-induced sensitization of transient receptor potential vanilloid 1 (TRPV1) channels. Endothelin-1 (ET1) released from endothelial cells, macrophages, mast cells, or keratinocytes can activate endothelin type A receptors (ETA) on nociceptive terminals. ETA receptor activation initiates a G-protein linked signaling cascade, in which the production of diacyl-glycerol (DAG) activates protein kinase C (PKC) ε. The latter phosphorylates TRPV1 channels. TRPV1 channel phosphorylation results in its activation, an influx of calcium, the depolarization of the resting membrane potential, and an increase in cell excitability and vesicle release of substance P. PLC: phospholipase C; IP3: inositol trisphosphate.
The detailed mechanisms of how ETA receptors trigger TRPV1 channel activation at peripheral terminals remain to be fully defined, but they may be related to calcium, an intracellular second messenger. Some studies report that ET-1 binding to ETA receptors increases the intracellular calcium concentration [81]. In substance P containing terminals in the spinal cord, intracellular calcium increases following ET-1 activation of dihydropyridine-sensitive calcium channels. Substance P, a sensitizer of primary afferent nociceptors, is then released from these terminals. In addition, ET-1-induced increases in intracellular calcium were found to contribute to the activation of sensory neurons through activation of PKCε[82], which phosphorylates TRPV1 receptors [75,76,82]. Indeed, in a mouse model of SCD, PKCε was found to be elevated in spinal dorsal horn neurons. Based on these studies, ET-1 binding to ETA receptors could initiate a downstream pathway in which PKCε activation sensitizes TRPV1 channels. This mechanism, however, has not been fully studied in SCD mice, but the basal elevation of ET-1 in SCD patients suggests that ET-1 may contribute to SCDassociated pain in a TRPV1-dependent manner.
Conclusions and Future Directions
SCD-associated chronic pain affects a majority of SCD patients, and dramatically reduces their quality of life. Uncovering the mechanisms of SCD-associated pain relies on our current understanding of chronic pain mechanisms, in addition to the discovery of SCD specific abnormalities. Unfortunately, the mechanism is still inconclusive. The variation in disease severity and pain among SCD patients has proven a complication in defining SCD-associated chronic pain. Vascular dysfunction and inflammation associated with SCD propose novel pain inducers, but further investigation is necessary to identify targets for pain relief. More clinical surveys exploring pain among SCD patients with defined SCD pain and its unique characteristics may help to unfold its complicity. Recent observations of possible social and environmental factors with molecular pathology may provide insight into pain variation among SCD patients. Future research should explore the unique pathophysiology of SCD associated pain including ET-1, which has grave potential as a possible target for pain intervention. Once again, understanding these mechanisms is essential to discovering effective novel therapies that are specific for SCD-associated pain, and researches need to be focusing this particular field.
Conflict Interests Disclosure
The authors have no conflicting interests to disclose.
References
- Hassell KL. Population Estimates of Sickle Cell Disease in the U.S. American Journal of Preventive Medicine 2010;38: S512-S521.
- Steiner, C and Miller, J. Sickle Cell Disease Patients in U.S. Hospitals, 2004. HCUP Statistical Brief #21. 2014. Rockville, MD, Agency for Healthcare Research and Quality.
- Kanter J, Kruse-Jarres R. Management of sickle cell disease from childhood through adulthood. Blood Reviews 2013;27:279-87.
- Bunn HF. Pathogenesis and Treatment of Sickle Cell Disease. N Engl J Med 1997;337:762-9.
- Ashley-Koch A, Yang Q, Olney RS. Sickle Hemoglobin (Hb S) Allele and Sickle Cell Disease: A HuGE Review. American Journal of Epidemiology 2000;151:839-45.
- Serjeant GR. The Natural History of Sickle Cell Disease. Cold Spring Harbor Perspectives in Medicine 2013;3.
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in Sickle Cell Disease. Rates and Risk Factors. N Engl J Med 1991;325:11-6.
- Vercellotti GM, Belcher JD. Not simply misshapen red cells: multimolecular and cellular events in sickle vasoocclusion. J Clin Invest 2014;124:1462-5.
- Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005;79: 17-25.
- Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. ASH Education Program Book 2013;2013:362-9.
- Solovey AAS. Modulation of endothelial cell activation in sickle cell disease: a pilot study. Blood 2001;97:1937-41.
- Solovey A, Kollander R, Shet A, et al. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 2004;104:840-6.
- Ellison AM, Shaw K. Management of Vasoocclusive Pain Events in Sickle Cell Disease. Pediatric Emergency Care 2007;23.
- Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily Assessment of Pain in Adults with Sickle Cell Disease. Annals of Internal Medicine 2008;148:94-101.
- Thompson WE, Eriator I. Pain Control in Sickle Cell Disease Patients: Use of Complementary and Alternative Medicine. Pain Med 2014;15:241-6.
- Nolan VG, Zhang Y, Lash T, et al. Association between wind speed and the occurrence of sickle cell acute painful episodes: results of a case-crossover study. Br J Haematol 2008;143:433-8.
- Rogovik AL, Persaud J, Friedman JN, et al. Pediatric Vasoocclusive Crisis and Weather Conditions. The Journal of Emergency Medicine 2011;41:559-65.
- Jacob E, Chan VW, Hodge C, et al. Sensory and Thermal Quantitative Testing in Children With Sickle Cell Disease. Journal of Pediatric Hematology/Oncology 9000; Publish Ahead of Print.
- Brandow AM, Stucky CL, Hillery CA, et al. Patients with sickle cell disease have increased sensitivity to cold and heat. Am J Hematol 2013;88:37-43.
- Telfer P, Bahal N, Lo A, Challands J. Management of the acute painful crisis in sickle cell diseasea re-evaluation of the use of opioids in adult patients. Br J Haematol 2014.
- Ballas SK. Current Issues in Sickle Cell Pain and Its Management. ASH Education Program Book 2007;2007:97-105.
- Ballas SK, Bauserman RL, McCarthy WF, et al. Hydroxyurea and Acute Painful Crises in Sickle Cell Anemia: Effects on Hospital Length of Stay and Opioid Utilization During Hospitalization, Outpatient Acute Care Contacts, and at Home. Journal of Pain and Symptom Management 2010;40:870-82.
- Chu LF, Angst MS, Clark D. Opioid-induced Hyperalgesia in Humans: Molecular Mechanisms and Clinical Considerations. The Clinical Journal of Pain 2008;24.
- Weber ML, Chen C, Li Y, et al. Morphine stimulates platelet-derived growth factor receptor-β signalling in mesangial cells in vitro and transgenic sickle mouse kidney in vivo. British Journal of Anaesthesia 2013;111:1004-12.
- Mihir Gupta, Lilian Msambichaka, Samir K B, Kalpna Gupta. Morphine for the Treatment of Pain in Sickle Cell Disease. The Scientific World Journal 2014;2015.
- Yawn BP, Buchanan GR, Afenyi-Annan AN. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033-48.
- Sheth S, Licursi M, Bhatia M. Sickle cell disease: time for a closer look at treatment options? Br J Haematol 2013;162:455-64.
- Ghasemi A, Keikhaei B, Ghodsi R. Side effects of hydroxyurea in patients with Thalassemia major and thalassemia intermedia and sickle cell anemia. Iran J Ped Hematol Oncol 2014;4:114-7.
- Okam MM, Ebert BL. Novel approaches to the treatment of sickle cell disease: the potential of histone deacetylase inhibitors. Expert Review of Hematology 2012;5:303-11.
- Molokie RE, Wilkie DJ, Wittert H, et al. Mechanismdriven phase I translational study of trifluoperazine in adults with sickle cell disease. European Journal of Pharmacology 2014;723:419-24.
- Wang ZJ, Wilkie DJ, Molokie R. Neurobiological Mechanisms of Pain in Sickle Cell Disease. ASH Education Program Book 2010;2010:403-8.
- Lu K, Cheng MCJ, Ge X, et al. A Retrospective Review of Acupuncture Use for the Treatment of Pain in Sickle Cell Disease Patients: Descriptive Analysis From a Single Institution. The Clinical Journal of Pain 2014;30.
- Schatz J, Schlenz A, McClellan CB, et al. Changes in Coping, Pain and Activity following CognitiveBehavioral Training: A Randomized Clinical Trial for Pediatric Sickle Cell Disease using Smartphones. The Clinical Journal of Pain 9000;Publish Ahead of Print.
- Tinti G, Somera R Jr, Valente FM, Doningos CR. Benefits of kinesiotherapy and aquatic rehabilitation on sickle cell anemia. A case report. Genet Mol Res 2010;9:360-4.
- Faes C, Balayssac-Siransy E, Connes P, et al. Moderate endurance exercise in patients with sickle cell anaemia: effects on oxidative stress and endothelial activation. Br J Haematol 2014;164:124-30.
- Machado RF, Kyle Mack A, Martyr S, et al. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol 2007;136:319-25.
- de Lima-Filho NN, Figueiredo MS, Vicari P, et al. Exercise-Induced Abnormal Increase of Systolic Pulmonary Artery Pressure in Adult Patients With Sickle Cell Anemia: An Exercise Stress Echocardiography Study. Echocardiography 2014.
- Tripette J, Hardy-Dessources MD, Romana M, et al. Exercise-related complications in sickle cell trait. Clinical Hemorheology and Microcirculation 2013;55:29-37.
- Connes P, Machado R, Hue O, Reid H. Exercise limitation, exercise testing and exercise recommendations in sickle cell anemia. Clinical Hemorheology and Microcirculation 2011;49:151-63.
- Cain DM, Vang D, Simone DA, et al. Mouse models for studying pain in sickle disease: effects of strain, age, and acuteness. Br J Haematol 2012;156:535-44.
- Pászty C, Brion CM, Manci E, et al. Transgenic Knockout Mice with Exclusively Human Sickle Hemoglobin and Sickle Cell Disease. Science 1997;278:876-8.
- Ryan TM, Ciavatta DJ, Townes TM. KnockoutTransgenic Mouse Model of Sickle Cell Disease. Science 1997;278:873-6.
- Manci EA, Hillery CA, Bodian CA, et al. Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood 2005;107:1651-8.
- Ryan TM, Townes TM, Reilly MP, et al. Human sickle hemoglobin in transgenic mice. Science 1990;247:566-8.
- Belcher JD, Bryant CJ, Nguyen J, et al. Transgenic sickle mice have vascular inflammation. Blood 2003;101:3953-9.
- Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: Modulation by cannabinoids. Blood 2014;116:456-65.
- Kenyon N, Wang L, Spornick N, et al. Sickle cell disease in mice is associated with sensitization of sensory nerve fibers. Experimental Biology and Medicine 2014.
- Zappia KJ, Garrison SR, Hillery CA, Stucky CL. Cold hypersensitivity increases with age in mice with sickle cell disease. PAIN. 2014.
- Latremoliere A, Woolf CJ. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. The Journal of Pain 2009;10:895-926.
- Garrison S, Kramer A, Gerges N, et al. Sickle cell mice exhibit mechanical allodynia and enhanced responsiveness in light touch cutaneous mechanoreceptors. Molecular Pain 2012;8:62.
- Cataldo G, Rajput S, Gupta K, Simone DA. Sensitization of nociceptive spinal neurons contributes to pain in a transgenic model of sickle cell disease. PAIN 2015.
- Hu HJ, Gereau RW. ERK Integrates PKA and PKC Signaling in Superficial Dorsal Horn Neurons. II. Modulation of Neuronal Excitability. Journal of Neurophysiology 2003;90:1680-8.
- Vincent L, Vang D, Nguyen J, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood 2013;122:1853-62.
- Steinhoff M, Vergnolle N, Young SH, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med 2000;6:151-8.
- Li D, Ren Y, Xu X, et al. Sensitization of Primary Afferent Nociceptors Induced by Intradermal Capsaicin Involves the Peripheral Release of Calcitonin GeneRelated Peptide Driven by Dorsal Root Reflexes. The Journal of Pain 2008;9:1155-68.
- Michaels LA, Ohene-Frempong K, Zhao H, Douglas SD. Serum Levels of Substance P Are Elevated in Patients With Sickle Cell Disease and Increase Further During Vaso-Occlusive Crisis. Blood 1998;92:3148-51.
- Teodoro FC, Tronco Júnior MF, Zampronio AR, et al. Peripheral substance P and neurokinin-1 receptors have a role in inflammatory and neuropathic orofacial pain models. Neuropeptides 2013;47:199-206.
- Hans G, Deseure K, Adriaensen H. Endothelin1-induced pain and hyperalgesia: A review of pathophysiology, clinical manifestations and future therapeutic options. Neuropeptides 2008;42:119-32.
- Barr TP, Kam S, Khodorova A, et al. New perspectives on the endothelin axis in pain. Pharmacological Research 2011;63:532-40.
- Ehrenreich H, Anderson RW, Fox CH, et al. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. The Journal of Experimental Medicine 1990;172:1741-8.
- Prasad VS, Palaniswamy C, Frishman WH. Endothelin as a Clinical Target in the Treatment of Systemic Hypertension. Cardiology in Review 2009;17.
- An SJ, Boyd R, Wang Y, et al. Endothelin-1 expression in vascular adventitial fibroblasts. American Journal of Physiology Heart and Circulatory Physiology 2006;290:H700-H708.
- Earley SN. Hypoxia-induced pulmonary endothelin-1 expression is unaltered by nitric oxide. Journal of Applied Physiology 2002;92:1152-8.
- Graido-Gonzalez E, Doherty JC, Bergreen EW, et al. Plasma Endothelin-1, Cytokine, and Prostaglandin E2Levels in Sickle Cell Disease and Acute VasoOcclusive Sickle Crisis. Blood 1998;92:2551-5.
- Gokin AP, Fareed MU, Pan HL, et al. Local Injection of Endothelin-1 Produces Pain-Like Behavior and Excitation of Nociceptors in Rats. The Journal of Neuroscience 2001;21:5358-66.
- Namer B, Hilliges M, Orstavik K, et al. Endothelin1 activates and sensitizes human C-nociceptors. PAIN 2008;137:41-9.
- Hans G, Schmidt BL, Strichartz G. Nociceptive sensitization by endothelin-1. Brain Research Reviews 2009;60:36-42.
- Hans G, Deseure K, Robert D, De Hert S. Neurosensory changes in a human model of endothelin-1 induced pain: A behavioral study. Neuroscience Letters 2007;418:117-21.
- Motta EM, Chichorro JG, Rae GA. Role of ETA and ETB endothelin receptors on endothelin-1-induced potentiation of nociceptive and thermal hyperalgesic responses evoked by capsaicin in rats. Neuroscience Letters 2009;457:146-50.
- Balonov K, Khodorova A, Strichartz GR. Tactile Allodynia Initiated by Local Subcutaneous Endothelin-1 Is Prolonged by Activation of TRPV-1 Receptors. Experimental Biology and Medicine 2006;231:1165-70.
- Pomonis JD, Rogers SD, Peters CM, et al. Expression and Localization of Endothelin Receptors: Implications for the Involvement of Peripheral Glia in Nociception. The Journal of Neuroscience 2001;21:999-1006.
- Khodorova A, Montmayeur JP, Strichartz G. Endothelin Receptors and Pain. The Journal of Pain 2009;10:4-28.
- Yamamoto T, Ono K, Hitomi S, et al. Endothelin Receptor-mediated Responses in Trigeminal Ganglion Neurons. Journal of Dental Research 2013;92:335-9.
- Zhou Z, Davar G, Strichartz G. Endothelin-1 (ET-1) Selectively Enhances the Activation Gating of Slowly Inactivating Tetrodotoxin-Resistant Sodium Currents in Rat Sensory Neurons: A Mechanism for the PainInducing Actions of ET-1. The Journal of Neuroscience 2002;22:6325-30.
- Hillery CA, Kerstein PC, Vilceanu D, et al. Transient receptor potential vanilloid 1 mediates pain in mice with severe sickle cell disease. Blood 2011;118:3376-83.
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov 2007;6:357-72.
- Belcher JD, Marker PH, Weber JP, et al. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood 2000;96:2451-9.
- Plant TD, Zöllner C, Mousa SA, Oksche A. Endothelin-1 Potentiates Capsaicin-Induced TRPV1 Currents Via the Endothelin A Receptor. Experimental Biology and Medicine 2006;231:1161-4.
- Montmayeur JP, Barr TP, Kam SA, et al. ET-1 induced Elevation of intracellular calcium in clonal neuronal and embryonic kidney cells involves endogenous endothelin-A receptors linked to phospholipase C through Gαq/11. Pharmacological Research 2011;64:258-67.
- Yamamoto H, Kawamata T, Ninomiya T, et al. Endothelin-1 enhances capsaicin-evoked intracellular Ca2+ response via activation of endothelin a receptor in a protein kinase Cε-dependent manner in dorsal root ganglion neurons. Neuroscience 2006;137:949-60.