Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/040
REVIEW ARTICLE OPEN ACCESS
The CRISPR/Cas9 system for gene editing and its potential application in pain research
Linlin Sun1, Brianna Marie Lutz2, Yuan-Xiang Tao1,2,3
1Department of Anesthesiology, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ 07103, USA
2Rutgers Graduate School of Biomedical Sciences, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ 07103, USA
3Departments of Cell Biology & Molecular Medicine, Physiology, Pharmacology, & Neuroscience, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ 07103, USA
Dr. Yuan-Xiang Tao, Department of Anesthesiology, New Jersey Medical School, Rutgers, The State University of New Jersey, 185 S. Orange Ave., MSB, E-661, Newark, NJ 07103. Tel: +1-973-972-9812; Fax: +1-973-972-1644, E-mail: yuanxiang.tao@njms.rutgers.edu
Editor: Renyu Liu, MD; PhD; Associate Professor, irector of Preoperative Medicine, Department of Anesthesiology and Critical Care; erelman School of Medicine at the University of Pennsylvania, 336 John Morgan building, 3620 Hamilton Walk, Philadelphia, PA 19104, Phone: 2157461485; FAX: 2153495078, Email: liur@uphs.upenn.edu
Received: June 16, 2016 | Accepted: July 08, 2016 | Published: July 21, 2016
Citation: Sun LL, Lutz BM, Tao YX. The CRISPR/ Cas9 system for gene editing and its potential application in pain research. Transl Perioper & Pain Med 2016; 1(3): 22-32.
Abstract
The CRISPR/Cas9 system is a research hotspot in genome editing and regulation. Currently, it is used in genomic silencing and knock-in experiments as well as transcriptional activation and repression. This versatile system consists of two components: a guide RNA (gRNA) and a Cas9 nuclease. Recognition of a genomic DNA target is mediated through base pairing with a 20-base gRNA. The latter further recruits the Cas9 endonuclease protein to the target site and creates double-stranded breaks in the target DNA. Compared with traditional genome editing directed by DNA-binding protein domains, this short RNA-directed Cas9 endonuclease system is simple and easily programmable. Although this system may have off-target effects and in vivo delivery and immune challenges, researchers have employed this system in vivo to establish disease models, study specific gene functions under certain disease conditions, and correct genomic information for disease treatment. In regards to pain research, the CRISPR/ Cas9 system may act as a novel tool in gene correction therapy for pain-associated hereditary diseases and may be a new approach for RNA-guided transcriptional activation or repression of pain-related genes. In addition, this system is also applied to loss-of-function mutations in pain-related genes and knockin of reporter genes or loxP tags at pain-related genomic loci. The CRISPR/Cas9 system will likely be carried out widely in both bench work and clinical settings in the pain field.
Keywords
CRISPR/Cas9, guide RNA, genomic editing, pain, gene correction therapy
Introduction
The CRISPR/Cas9 system is becoming increasingly popular in the field of genomic editing and gene regulation. This system was developed based on the RNA-guided Cas9 endonuclease found in bacteria immune system. CRISPR refers to clustered regularly interspaced short palindromic repeats, which are segments of prokaryotic DNA containing short repeats of base sequences. Each repetition is followed by short sequence of "spacer DNA" derived from previous virus intruder genome [1]. Subsequent transcriptions from the CRISPR repeat-spacer units yield two noncoding RNAs: one CRISPR RNA (crRNA) containing nuclease guide sequences compiled from the spacers, and another noncoding RNA, complementary to the repeat sequence, known as trans-activating crRNA (tracrRNA). When a virus invades the bacterium for a second time, this dual crRNA:tracrRNA is now capable of recognizing the viral DNA as foreign, and thus base pairs with the intruder DNA and recruits Cas9 endonuclease to create double strand DNA (dsDNA) breaks at the recognition site, ultimately this leads to silencing of the viral gene [1-3]. The CRISPR/Cas9 system is the type II prokaryotic immune system, which provides acquired immunity through gaining resistance to foreign genetic elements [1]. By delivering the Cas9 endonuclease and appropriate guide RNAs into mammalian system, genome editing in mammalian cells could be efficiently performed.
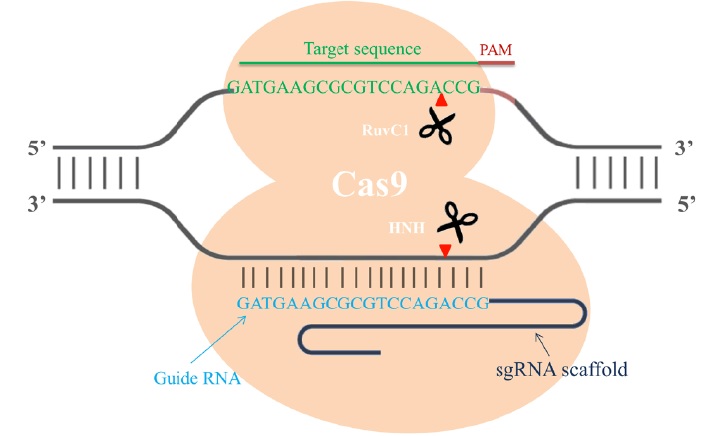
Cas9 is an endonuclease with two enzymatic domains: an HNH domain that cleaves the complementary strand of DNA that base pairs with the guide RNA and an RuvC1 domain that cleaves the noncomplementary strand [4] (Fig. 1). The RNA-guided Cas9 system will only recognize its target sequence if that target sequence is immediately followed by a protospacer-adjacent motif (PAM) sequence at the 5' end [5] (Fig. 1). To improve the efficiency of the CRISPR/Cas9 system in genome editing, recent studies designed single chimeric guide RNA (sgRNA) molecules. These molecules contain a target recognition 20-base RNA sequence mimicking the function of crRNA, followed by a hairpin scaffold structure mimicking the base-pairing interactions between tracrRNA and crRNA [3](Fig. 1). Once dsDNA breaks are achieved in the targeted gene, cells activate their error-prone non-homologous end joining repair (NHEJ) pathways to fix the damage, resulting in random insertion/deletion mutations (indels) of DNA bases at the cut site. Introduction of indels to the coding frame of the targeted gene consequently leads to changes in the target gene expression including genetic knockdown [5] (Fig. 2). If a homologous DNA template is provided, cells repair their DNA through homologous recombination, resulting in genomic knock-in at the specific cut site (Fig. 2). Thus, the CRIPSR/Cas9 system can be used in mammalian genomic editing including the production of knockout or knockin models.
Figure 1: Single chimeric RNA-guided Cas9 endonuclease recruitment for target recognition. Single chimeric guide RNA (blue) directs the active Cas9 endonuclease (orange) to cleave site-specific DNA when the targeted sequence (green) is immediately followed by a PAM sequence (red). The RuvC1 domain of Cas9 cleaves the noncomplementary strand, and the HNH domain cleaves the complementary strand of DNA. The cut site usually occurs 3 bp upstream of the PAM sequence (red arrow).
Genomic Editing
Disruption of a target gene is a way to decipher the function of a gene. As discussed above (Fig. 2), by using the CRIPSR/Cas9 system, genomic editing can be achieved by creating a single gRNA directed toward the gene of interest that will recruit a Cas9 endonuclease. This versatile system has been carried out to disrupt several genes in order to decipher their functions [6] or to provide a potential gene correction therapy in mammals [7,8]. For example, the disruption of CDK11 was achieved in an osteosarcoma cell line using the CRISPR/Cas9 system, resulting in decreases in cell proliferation and viability [6]. The human pcsk9 (proprotein convertase subtilisin/kexin type 9) gene has emerged as a promising therapeutic target for cardiovascular disease, given that individuals with naturally occurring loss-of-function pcsk9 mutations experience reduced low-density lipoprotein cholesterol levels which protects against cardiovascular disease. Achieving loss of function mutations in the Pcsk9 gene using the CRISPR/Cas9 system successfully decreased serum PCSK9 by 90% and total cholesterol levels by 40% in a mouse model [7,9]. Moreover, by introducing multiple gRNAs simultaneously, this system could easily be adapted to target multiple genes at the same time. This may be important for studying the effects of a family of proteins or a pathway on behaviors. The role of DNA methylation in the development of human embryonic stem cells was investigated through successful co-knockout of the three key DNA methyltransferase enzymes, DNMT1, DNMT3a and DNMT3b by co-application of three gRNAs targeting each enzyme [10]. Commercially available CRISPR gRNA libraries offer a convenient approach to achieve large-scale loss-of-functionbased screening in different species. By knocking out thousands of genes individually using a genome-scale mouse CRISPR gRNA library in a non-metastatic cancer model, the top metastatic cancer contributors were dissected [11]. Another gRNA library targeting 19,150 mouse protein-coding genes was designed to reveal their roles in drug resistance [12].
Figure 2: Genomic editing. Once double strand DNA breaks are achieved, cells activate their error-prone non homologous end joining (NHEJ) repair pathways to fix the damage by introducing random small insertions or deletions at the cut site. However, if a homologous DNA template is provided, cells repair their DNA by homologous recombination (HR), resulting in genomic knock-in at the specific cut site.
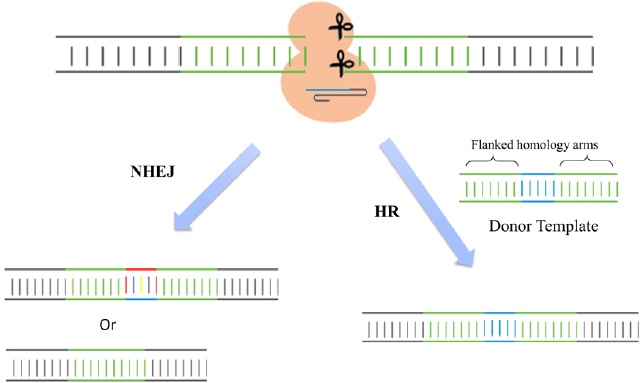
The CRISPR/Cas9 system has also been used for specific gene knockin studies. As described in Figure 2, the cut at the locus of interest is induced by gRNA-directed Cas9 endonuclease. When a DNA cassette flanked by homology arms is provided as a donor template, the cut site is repaired through homologous recombination and the template is incorporated into the selective DNA cassette [13] (Fig. 2). Since the CRISPR targeting site is destroyed by gene insertion, this CRISPR-Cas9 system is mostly carried out to enable reporter gene knockin for the identification and purification of specific cell types of interest. With the use of this system, researchers have successfully generated mice carrying V5 tags, mCherry or GFP fluorescence reporters at the 3' ends of target genes or insertion of two loxP sites flanking an exon of a target gene [14]. Similar engineering was also carried out in rats to obtain reporter knockin and floxed alleles [15,16]. The principles established in these studies could directly apply to other species for simplifying the genome engineering process.
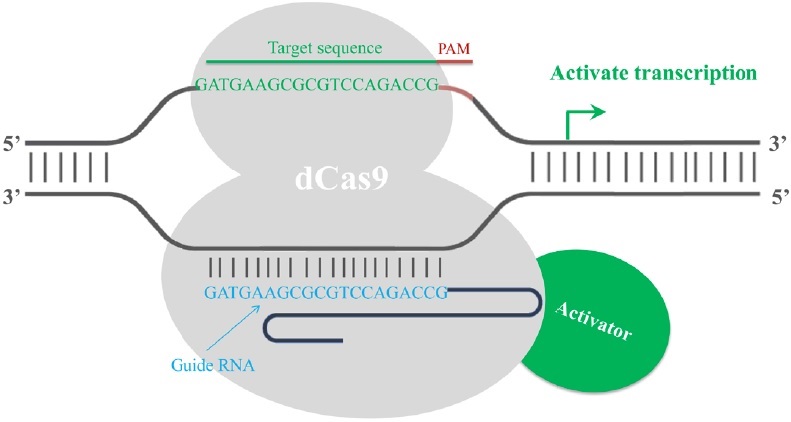
Finally, the CRISPR/Cas9 system has been employed in transcription regulation. The gRNA-mediated recruitment of a cleavage inactive form of Cas9 [dead Cas9 (dCas9), D10A mutant in RuvC1 domain and H841A mutant in HNH domain] enables the gRNA-dCas9 complex to harbor specific regulatory positions of a given gene, acting as a scaffold to recruit transcriptional regulators and interfere with transcriptional elongation, RNA polymerase binding or transcription factor binding, resulting in alternations in the expression of target genes [17]. Fusion of dCas9 with a transcriptional repressor domain robustly silences the endogenous target genes. For example, dCas9 has been fused to KRAB (Krüppel-associated box, a category of repressive chromatin modifier domains) [18], which resulted in transcription repression (Fig. 3). In contrast, dCas9 can be converted into an RNA-guided transcription activator (dCas9-activator) when fused to transcriptional activation domains [19,20], as in the case of dCas9-VP64 (virus protein 64, tetrameric virus protein 16 transcription activator domain) (Fig. 4). In order to obtain highly specific and efficient transcriptional regulation, the design of the fusion protein or fusion partner is key for future optimization efforts.
Figure 3: RNA-mediated recruitment of the dCas9transcriptional repressor domain interferes with the transcription process. A dead Cas9 protein (grey) is fused with a transcriptional repressor domain (red). The transcriptional repressor (red) is then recruited to the target site by gRNA-dCas9 and silences the transcription of the target gene. The repression is most efficient if the target site is within a 200 bp window upstream of the transcription start site.
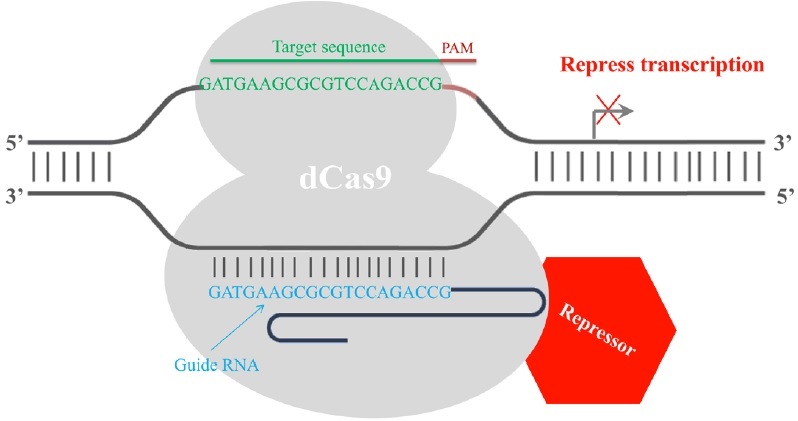
Figure 4: Fusion of dCas9 with a transcriptional activation domain acts as a RNA-guided transcription activator. A dead Cas9 protein (grey) is fused with a transcriptional activator domain (green). The transcriptional activator (red) is then recruited to the target site by gRNA-dCas9 and activates the transcription of the target gene. The activation is most efficient if the target site is within a 200 bp window upstream of the transcription start site.
Advantages over Other Methods
Currently, ZFN, TALEN and CRISPR/Cas9 system, comprise a powerful class of tools for genomic engineering. Zinc finger nucleuses are artificial chimeric site-specific endonuclease engineered by fusing a zinc finger DNA-binding domain to a non-specific DNA-cleavage domain of the restriction endonuclease FokI [21,22]. The zinc finger DNA binding domain consists of 3-6 zinc finger units, with each unit recognizing 3 base-pairs of DNA. Since the non-specific nuclease FokI only creates a dsDNA break when it dimerizes, ZFN targets consist of two zinc-finger binding sites separated by a 5–7-bp spacer sequence recognized by the FokI cleavage domain (Fig. 5A). Once the cut is achieved, the NHEJ pathway takes place and results in loss of gene function [21,22]. However, the various zinc finger units when assembled in arrays interfere with each other, resulting in decreased efficiency in binding the DNA targets [23,24]. With the necessity of dimerization of the system, the ZFN system has accessible targets approximately every 500 basepairs of random genomic sequence [25,26], while CRISPR/Cas9 has a targeting range of 1 in every 8 basepairs of random genomic sequence [27]. Due to this limitation in the sequences recognized by ZFNs, the improved artificial nuclease TALEN, similar to ZFN, was introduced. TALEN is composed of one transcription-like effector (TALE) DNA binding domain, consisting of ~ 500-700 amino acids, and also one non-specific DNA cleavage domain [28-30] (Fig.5B). Based on the already deciphered DNA:protein binding codes [31,32], TALE domain now can be designed to recognize almost any desired sequence.
Figure 5: Protein-DNA interactions direct dsDNA breaks mediated through dimerized FokI nucleases in ZFN (A) and TALEN (B) systems. A. Each zinc finger nuclease consists of a DNA-binding domain and a non-specific DNA-cleavage domain of the restriction endonuclease FokI (grey). The DNA-binding domain is composed of 3-6 zinc finger units (colored), with each recognizing 3 bp of DNA sequence. The dsDNA cleavage requires dimerization of two FokI nuclease domains. Therefore, each ZFN target consists of two zinc-finger binding sites separated by a spacer sequence recognized by the FokI cleavage domain. B. TALEN is also composed of one DNA binding domain and one non-specific DNA cleavage domain FokI. The DNA binding domain is engineered according to the already deciphered DNA:protein binding codes (red, green, yellow and blue-indicated amino acid sequences). Each TALEN target also consists of two recognition sites separated by a spacer sequence recognized by the FokI cleavage domain, which creates a dsDNA break if Fok1 dimerizes.
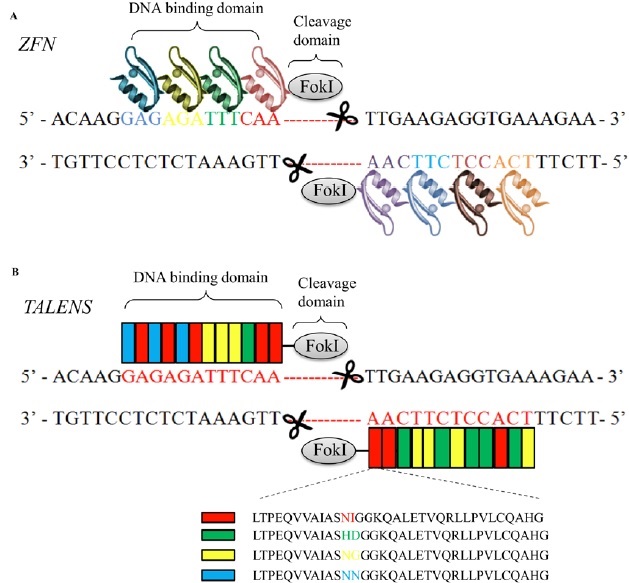
However, both nuclease domains in ZFN and TALEN systems recognize their genomic targets by amino acid sequence in the DNA binding domain of either zinc finger protein or TALE protein (protein-DNA interaction), which means the amino acid sequence of their DBD has to be redesigned for each target site, which can be a painstakingly lengthy process. In contrast, Cas9 endonuclease-mediated dsDNA breaks only require co-expression of a single target-specific guide RNA. The specificity of ZFN and TALEN can be enhanced by increasing the number of zinc finger or TALE modules [33], whereas the specificity of CRISPR-Cas9 is dependent on a 20 bp guide RNA. Moreover, since the targeting specificity of ZFN and TALEN is defined solely by protein-DNA interactions, their off-targets are context-dependent and cannot be completely predicted based on the DNA sequence homology. On the contrary, CRISPR-Cas9 recognizes targets based on Watson-Crick basepairing rules. Therefore, the off-target sites in the CRISPR-Cas9 system can be more reliably predicted based on sequence homology. Taken together, compared to the traditional genome editing tools, the CRISPR/Cas9 system has several advantages including simplicity in target design, genome-wide accessibility, the ability to target multiple sites in one step, and predictable off-target effects.
Prospective applications in pain research and treatments
Based on the characteristics of the CRIPSR/Cas9 system in genomic editing and its advantages over traditional genomic editing strategies described above, it is expected that this system may have potential applications in the generation of pain-related transgenic animals, the exploration of chronic pain mechanisms, and the treatment of pain-associated congenital diseases.
Genetically modified mice created using various transgenic technologies have been widely used in preclinical pain research. Conventional or congenital genetic KO mice have been generated through gene targeting with homology recombination in embryonic stem cells [34]. Using this approach, researchers have studied the functions of a pain-related gene in vivo. However, given that the gene is deleted from conception, this model has several limitations. As a whole-body KO, this model lacks the ability to identify tissue-specific effects of gene deficiency. Some gene KO strains die in early developmental stages and this prevents researchers from studying the function of these genes through adulthood. Moreover, knocking out a target gene during the embryonic period may result in compensatory changes in the expression of other genes or non-specific changes during the development that may affect animal behaviors [35,36]. The generation of conditional genetic KO mice with spatial and temporal control of gene inactivation using the Cre-loxP system may avoid these limitations. However, the fertility rate of tamoxifen-inducible Advillin-Cre-ERT2 recombinase mice [37] that are presently available for studying the role of individual genes in adult sensory neuron function is rather low (personal communications with Dr. John N Wood's Lab). In addition, other genetically modified animals (except for transgenic mice) are unavailable because germline-competent embryonic stem cells were achieved only in mice. The CRISPR/Cas9 system may overcome the limitations caused by these traditional transgenic technologies. This system is able to create both germline and somatic animal models with point mutations, deletions and complex chromosomal rearrangements [38]. Indeed, recent researchers have carried out the easily programmable CRISPR/Cas9 system to generate genetically modified mouse and rat lines [14-16]. The principles established in these studies could directly be applied to pain-related genes. In addition, given that the CRISPR/Cas9 system is simple in target design and has wide genome accessibility and predictable off-target effects, genome engineering using this system could be less time consuming.
Chronic pain is an unpleasant, long-term sensory and emotional experience that affects millions of people worldwide. Unsatisfactory effective treatment for this disorder is partially due to a lack of knowledge concerning the molecular mechanisms that underlie chronic pain development and maintenance. In the past decades, several pathophysiological mechanisms have been proposed. For example, peripheral nerve injury-induced abnormal ectopic firing in the neuroma at the injured site and dorsal root ganglion neurons is thought to contribute to nerve injury-induced pain hypersensitivity [39-42]. The occurrence of such ectopic firing may be related to the reduced expression of some voltage-gated potassium channels (Kvs) such as Kv1.2 in DRG [43-46]. In addition, the expression of opioid receptors in the injured dorsal root ganglion (DRG) significantly decreased following peripheral nerve injury. This decrease not only results in reduced opioid analgesia in neuropathic pain management, but also enhanced release of neurotransmitters in primary afferents which participates in neuropathic pain genesis [47,48]. Thus, rescuing the reduction in these receptors and/or channels may attenuate the development and maintenance of neuropathic pain [43]. Using the CRISPR/Cas9 system, designing a sequence specific guide RNA targeting the genes encoding opioid receptors and voltage-gated potassium channels (e.g., Kv1.2), combined with an RNA-guided nuclease-deficient Cas9 (dCas9) protein fused with transcriptional activators, could rescue the reductions of these pain-related genes through their enhanced transcription and alleviate nerve injury-induced pain hypersensitivity. Thus, the application of the CRISPR/Cas9 system not only further elucidates the mechanisms underlying chronic pain but also can be used as a complementary strategy for pharmacological drugs in the treatment of this disorder.
Patients with some congenital diseases display sensory changes that alter their perception of pain. Cohorts of patients suffer from hereditary sensory and autonomic neuropathies (HSAN) that result in a marked absence of pain sensitivity [49]. These patients lack itch sensations, deep pain sensations in bones and joints and, most importantly, the protective reflexes induced by pain perception [50]. HSAN type IV is linked to mutations in the gene encoding for the tropomyosin receptor kinase A, a nerve growth factor (NGF) receptor [51], whereas HSAN type V is associated with a mutation in the NGF gene [52]. Loss-of-function mutations in the SCN9A gene that encode voltage-gated sodium channel Nav1.7 result in congenital insensitivity to pain in humans and mice [53,54]. Gain-of-function mutations in the SCN9A gene result in spontaneous pain as observed in paroxysmal extreme pain disorder and primary erythermalgia [55-57]. Gain-of-function mutations in the SCN10A gene that encode voltage-gated sodium channel Nav1.8 also result in painful peripheral neuropathy [58]. Gain-of-function mutations in the SCN11A gene that encodes voltage-gated sodium channel Nav1.9 cause painful neuropathy [59-61] or insensitivity to pain [62,63] depending on the site and content being mutated. Therefore, the CRISPR/Cas9-meidiated homology directed repair systems guided by specific guide RNAs targeting these mutated genes may provide in vivo gene correction therapy for these congenital diseases.
Future challenges in the application
The CRISPR/Cas9 system has been used intensively in in vitro experiments, but its application in in vivo studies may have several challenges including off-target effects, in vivo delivery vectors, and immune responses.
Off-target effects of the CRISPR-Cas9 system usually result from mismatches between the 20-base guide RNA and its target DNA sequence. Of note, multiple mismatches could be tolerated depending on the quantity, position and base identity of mismatches. For example, within the 20-base guide RNA, mismatches are less tolerated in the second half because the second half recognizes the seed region (6-11 bp upstream of PAM) in the targeted DNA sequence, leading to more off-target effects [3,5]. Efforts have been made to minimize the CRISPR-Cas9's off-target effects. Cho, et al. reported that, by modifying guide RNA and Cas9, the system could effectively discriminate on-target sites from off-target sites [64]. Dual crRNA-tracrRNA rather than single chimeric guide RNA or the use of synthetic guide RNA instead of guide-RNA coding plasmids also help alleviate off-target effects [64]. The utility of single or dual Cas9 nickase could also significantly reduce off-target effects. In gain-of-function research, the utility of a mutant variant of Cas9 nickase (D10A in its RuvC I domain) that creates only single stranded breaks could significantly reduce off-target effects as nicked DNA tends to be repaired by high-fidelity homologous recombination [8,13,64]. In loss-of-function studies, implementation of dual Cas9 nickases on opposite DNA strands with separate guide RNAs leads to efficient dsDNA break formation with 50to 1500-fold fewer off-target insertions or deletions [65].
The viral vectors that express the CRISPR/Cas9 system for in vivo delivery may have safety concerns, limited packaging capacities, and limitations in the infected cell types. Although adenoviral, adeno-associated viral (AAV), and lentiviral vectors have been applied for delivery of the CRISPR/Cas9 system in vivo [12,66,67], AAV vectors are the most promising in vivo vehicles because they have little immunogenic potential or endogenous vector recombination [68] and a broad range of serotype specificity [69,70]. Additionally, the restrictive ~4.5 kb cargo size of AAV vectors could be overcome by packaging of a smaller-sized Staphylococcus aureus Cas9 (SaCas9) orthologue instead of the 4.2 kb S. pyogenes Cas9 (SpCas9) nuclease [9]. Recent advances in non-viral vehicles make them potential alternatives for CRISPR/Cas9 delivery. Compared with viral vectors, non-viral vehicles do not have viral safety concerns, and are generally easier to be synthesized or produced [71]. One such non-viral vehicle, a nanoparticle named "7C1", has been used for in vivo delivery of guide RNA in Cre-dependent and constitutive Cas9-expressing mice [72]. Further optimization in in vivo delivery vehicles will provide a potential use of the CRISPR/Cas9 system in translational research.
A recent study reported that Cas9 as a foreign bacterial protein led to an immune response in mammals [73]. Upon Cas9 protein stimulation in mice, Cas9-specific antibody was generated and interleukin-2 secreted from the Cas9-primed splenocytes [73]. Therefore, further efforts should be made in defining immune epitopes of Cas9 and modifying the endonuclease to limit its immunogenicity, in addition to monitoring Cas9-induced immune responses, in in vivo research.
Conclusion
The CRISPR/Cas9 technology is a revolutionizing approach in genome editing and regulation. Compared with previous genome editing methods, this short RNA-directed Cas9 nuclease system has several advantages including its simplicity in target design, wide genome accessibility, ability to target multiple sites in one step, and predictable off-target effects. Even if the application of the CRISPR/Cas9 system in in vivo studies has some challenges, researchers have begun to carry out this system to establish disease models, study specific gene functions under certain disorder conditions, and correct genomic information for disease treatment. With respect to pain research, the CRISPR/Cas9 system will likely offer a novel and less time consuming approach for gene correction therapy, gene expression regulation, and genetic engineering for animal lines. in vivo targeting or correcting of pain specific genes under chronic pain conditions by use of the CRISPR/Cas9 system might offer therapeutic targets for chronic pain management.
Author Disclosure
The authors declare no conflicts of interest.
Disclosure of Funding
This work was supported by grants from the National Institutes of Health, Bethesda, Maryland (grant numbers: NS072206, HL117684, and DA033390).
Conflict Interests Disclosure
The authors have no conflicting interests to disclose.
References
- Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012; 482.
- Bhaya D, Davison M, Barrangou R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annual Review Genetics, Vol 45 2011; 45.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012; 337.
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014; 156.
- Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013; 339.
- Feng Y, Sassi S, Shen JK, Yang XQ, Gao Y, Osaka E, et al. Targeting Cdk11 in Osteosarcoma Cells Using the CRISPR-cas9 System. Journal of Orthopaedic Research 2015; 33.
- Ding QR, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, et al. Permanent Alteration of PCSK9 With in vivo CRISPR-Cas9 Genome Editing. Circulation Research 2014; 115.
- Mali P, Yang LH, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-Guided Human Genome Engineering via Cas9. Science 2013; 339.
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. in vivo genome editing using Staphylococcus aureus Cas9. Nature 2015; 520.
- Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet 2015; 47: 469-478.
- Chen SD, Sanjana NE, Zheng KJ, Shalem O, Lee K, Shi X et al. Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 2015; 160.
- Koike-Yusa H, Li YL, Tan EP, Velasco-Herrera MD, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature Biotechnology 2014; 32.
- Merkle FT, Neuhausser WM, Santos D, Valen E, Gagnon JA, Maas K, et al. Efficient CRISPR-Cas9-Mediated Generation of Knockin Human Pluripotent Stem Cells Lacking Undesired Mutations at the Targeted Locus. Cell Reports 2015; 11.
- Wang HY, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013; 153: 910-918.
- Ma YW, Ma J, Zhang X, Chen W, Yu L, Lu YD, et al. Generation of eGFP and Cre knockin rats by CRISPR/Cas9. Febs Journal 2014; 281: 3779-3790.
- Ma YW, Shen B, Zhang X, Lu YD, Chen W, Ma J, et al. Heritable Multiplex Genetic Engineering in Rats Using CRISPR/Cas9. Plos One 2014; 9.
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013; 152.
- Gilbert LA, Larson MH, Morsut L, Liu ZR, Brar GA, Torres SE, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013; 154.
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014; 159.
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology 2013; 31.
- Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Molecular and Cellular Biology 2001; 21.
- Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nature Biotechnology 2005; 23.
- Lam KN, van Bakel H, Cote AG, van der Ven A, Hughes TR. Sequence specificity is obtained from the majority of modular C2H2 zinc-finger arrays. Nucleic Acids Research 2011; 39.
- Tan WS, Carlson DF, Walton MW, Fahrenkrug SC, Hackett PB. Precision editing of large animal genomes. Adv Genet 2012; 80.
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 2009; 19.
- Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 2008; 5.
- Ansai S, Kinoshita M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biology Open 2014; 3.
- Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science 2011; 333.
- Mashimo T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev Growth Differ 2014; 56.
- Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, et al. Targeted Genome Editing Across Species Using ZFNs and TALENs. Science 2011; 333.
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009; 326.
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science 2009; 326.
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nature Biotechnology 2012; 30.
- Mansour SL, Thomas KR, Capecchi MR. Disruption of the Proto-Oncogene Int-2 in Mouse Embryo-Derived Stem-Cells A General Strategy for Targeting Mutations to Non-Selectable Genes. Nature 1988; 336: 348-352.
- Tao F, Liaw WJ, Zhang B, Yaster M, Rothstein JD, Johns RA, et al. Evidence of neuronal excitatory amino acid carrier 1 expression in rat dorsal root ganglion neurons and their central terminals. Neuroscience 2004; 123: 1045-1051.
- O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, et al. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science 1994; 265: 542-546.
- Lau J, Minett MS, Zhao J, Dennehy U, Wang F, Wood JN, et al. Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible Advillin-Cre-ERT2 recombinase mouse. Mol Pain 2011; 7: 100.
- Mou HW, Kennedy Z, Anderson DG, Yin H, Xue W. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Medicine 2015; 7.
- Bird EV, Christmas CR, Loescher AR, Smith KG, Robinson PP, Black JA, et al. Correlation of Nav1.8 and Nav1.9 sodium channel expression with neuropathic pain in human subjects with lingual nerve neuromas. Molecular Pain 2013; 9.
- Dib-Hajj SD, Black JA, Waxman SG. Na-V 1.9: a sodium channel linked to human pain. Nature Reviews Neuroscience 2015; 16: 511-519.
- Shields SD, Cheng XY, Uceyler N, Sommer C, Dib-Hajj SD, Waxman SG. Sodium Channel Na(v)1.7 Is Essential for Lowering Heat Pain Threshold after Burn Injury. Journal of Neuroscience 2012; 32: 10819-10832.
- Xie WR, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel Na(V)1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 2013; 154: 1170-1180.
- Fan L, Guan X, Wang W, Zhao JY, Zhang H, Tiwari V, et al. Impaired neuropathic pain and preserved acute pain in rats overexpressing voltage-gated potassium channel subunit Kv1.2 in primary afferent neurons. Molecular Pain 2014; 10.
- Kim DS, Choi JO, Rim HD, Cho HJ. Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Molecular Brain Research 2002; 105: 146-152.
- Laumet G, Garriga J, Chen SR, Zhang YH, Li DP, Smith TM, et al. G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nature Neuroscience 2015; 18: 1746-1755.
- Park SY, Choi JY, Kim RU, Lee YS, Cho HJ, Kim DS. Downregulation of voltage-gated potassium channel alpha gene expression by axotomy and neurotrophins in rat dorsal root ganglia. Molecules and Cells 2003; 16: 256-259.
- Wei LN. Epigenetic control of the expression of opioid receptor genes. Epigenetics 2008; 3: 119-121.
- Zhou XL, Yu LN, Wang Y, Tang LH, Peng YN, Cao JL, et al. Increased methylation of the MOR gene proximal promoter in primary sensory neurons plays a crucial role in the decreased analgesic effect of opioids in neuropathic pain. Molecular Pain 2014; 10.
- Dyck PJ, Mellinger JF, Reagan TJ, Horowitz SJ, Mcdonald JW, Litchy WJ, et al. Not Indifference to Pain But Varieties of Hereditary Sensory and Autonomic Neuropathy. Brain 1983; 106: 373-390.
- Capsoni S. From genes to pain: nerve growth factor and hereditary sensory and autonomic neuropathy type V. European Journal of Neuroscience 2014; 39: 392-400.
- Indo Y. Molecular basis of congenital insensitivity to pain with anhidrosis (CIPA): Mutations and polymorphisms in TRKA (NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Human Mutation 2001; 18: 462-471.
- Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, Solders G, et al. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Human Molecular Genetics 2004; 13: 799-805.
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006; 444: 894-898.
- Staud R, Price DD, Janicke D, Andrade E, Hadjipanayis AG, Eaton WT, et al. Two novel mutations of SCN9A (Nav1.7) are associated with partial congenital insensitivity to pain. European Journal of Pain 2011; 15: 223-230.
- Dib-Hajj SD, Rush AM, Cummins TR, Hisama FM, Novella S, Tyrrell L, et al. Gain-of-function mutation in Na(v)1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005; 128: 1847-1854.
- Drenth JPH, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. Journal of Clinical Investigation 2007; 117: 3603-3609.
- Lee MJ, Yu HS, Hsieh ST, Stephenson DA, Lu CJ, Yang CC. Characterization of a familial case with primary erythromelalgia from Taiwan. Journal of Neurology 2007; 254: 210-214.
- Faber CG, Lauria G, Merkies ISJ, Cheng XY, Han CY, Ahn HS, et al. Gain-of-function Na(v)1.8 mutations in painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America 2012; 109: 19444-19449.
- Han CY, Yang Y, de Greef BTA, Hoeijmakers JGJ, Gerrits MM, Verhamme C, et al. The Domain II S4-S5 Linker in Nav1.9: A Missense Mutation Enhances Activation, Impairs Fast Inactivation, and Produces Human Painful Neuropathy. Neuromolecular Medicine 2015; 17: 158-169.
- Huang JY, Han CY, Estacion M, Vasylyev D, Hoeijmakers JGJ, Gerrits MM, et al. Gain-of-function mutations in sodium channel Na(V)1.9 in painful neuropathy. Brain 2014; 137: 1627-1642.
- Zhang XY, Wen JM, Yang W, Wang C, Gao LN, Zheng LH, et al. Gain-of-Function Mutations in SCN11A Cause Familial Episodic Pain. American Journal of Human Genetics 2013; 93: 957-966.
- Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nature Genetics 2013; 45: 1399-+.
- Leipold E, Hanson-Kahn A, Frick M, Gong P, Bernstein JA, Voigt M, et al. Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat Commun 2015; 6.
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Research 2014; 24.
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013; 154.
- Maggio I, Holkers M, Liu J, Janssen JM, Chen XY, Goncalves MAFV. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Scientific Reports 2014; 4.
- Senis E, Fatouros C, Grosse S, Wiedtke E, Niopek D, Mueller AK, et al. CRISPR/Cas9-mediated genome engineering: An adeno-associated viral (AAV) vector toolbox. Biotechnology Journal 2014; 9.
- Vasileva A, Jessberger R. Precise hit: Adeno-associated virus in gene targeting. Nature Reviews Microbiology 2005; 3.
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nature Reviews Genetics 2011; 12.
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy 2008; 16.
- Li L, He ZY, Wei XW, Gao GP, Wei YQ. Challenges in CRISPR/CAS9 Delivery: Potential Roles of Nonviral Vectors. Hum Gene Ther 2015; 26.
- Platt RJ, Chen SD, Zhou Y, Yim MJ, Swiech L, Kempton HR, et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014; 159.
- Wang D, Mou H, Li S, Li Y, Hough S, Tran K, et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum Gene Ther 2015; 26.
Table of Contents
- Abstract
- Keywords
- Introduction
- Genomic Editing
- Advantages over Other Methods
- Prospective applications in pain research and treatments
- Future challenges in the application
- Conclusion
- Author Disclosure
- Disclosure of Funding
- Conflict Interests Disclosure
- Figure 1
- Figure 2
- Figure 3
- Figure 4
- Figure 5
- References