Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/170
Case Report | Volume 10 | Issue 1 Open Access
An Anesthetic Management of Patient with King Denborough Syndrome and Susceptible Malignant Hyperthermia a Undergoing Tracheostomy: A Case Report
Oktom Nurumbetova, MD*, Ahmed Uslu, MD, Deniz Kaya, MD, Nedim Çekmen, MD, PhD and Zeynep Kayhan, MD, PhD
Department of Anesthesiology, Faculty of Medicine, Baskent University, Ankara, Turkey
Oktom Nurumbetova, MD, Department of Anesthesiology, Faculty of Medicine, Baskent University, Fevzi Cakmak Caddesi 10, Sokak No:45 Bahcelievler, 06490 Ankara, Turkey, Tel: +0312-203-68-68- 4867; +905528114689, E-mail: n.oktom.o@gmail.comEditor: Renyu Liu, MD; PhD; Professor, Department of Anesthesiology and Critical Care, Perelman School of Medicine at the University of Pennsylvania, Center of Penn Global Health Scholar, 336 John Morgan building, 3620 Hamilton Walk, Philadelphia, PA 19104, USA, Fax: 2153495078, E-mail: RenYu.Liu@pennmedicine.upenn.edu
Received: January 10, 2023 | Accepted: March 27, 2023 | Published: March 27, 2023
Citation: Nurumbetova O, Uslu A, Kaya D, Çekmen N, Kayhan Z. An Anesthetic Management of Patient with King Denborough Syndrome and Susceptible Malignant Hyperthermia a Undergoing Tracheostomy: A Case Report. Transl Perioper Pain Med 2023; 10(1):508-511
Abstract
King-Denborough syndrome (KDS) is a rare genetic syndrome expressed with myopathy, skeletal and craniofacial features, and malignant hyperthermia susceptibility (MHS) and affected patients with neuromuscular disorders (NMD) present variable clinical manifestations and implications for anesthesiologists. A 28-month-old preterm male infant diagnosed with KDS with congenital laryngomalacia and epilepsia underwent tracheostomy under general anesthesia performed by total intravenously (i.v.) anesthesia and no use of the neuromuscular blocking agents. There were no anesthesia-related complications during the intraoperative and postoperative periods. Thus, we report the successful anesthesia management of patients with structural myopathies and dysmorphic findings with NMD presents a seriously challenging and increasing awareness of anesthesia-related complications; however, more studies are needed for conclusive information.
Keywords
King-Denborough syndrome, Neuromuscular disorder, Malignant hyperthermia, Anesthesia management
Introduction
The King Denborough Syndrome (KDS) is an uncommon autosomal dominant disorder associated with Noonan-like features and an MHS [1]. This congenital disease is characterized by slowly progressive myopathy, craniofacial abnormalities such as low-set ears, malar hypoplasia, micrognathia, ptosis, down-slanting palpebral fissures, hypertelorism, short and webbed neck, epicanthic folds and skeletal deformities which includes thoracic kyphosis, lumbar scoliosis, pectus excavatum/carinatum, winging of the scapulae, palmar simian line [2,3]. The structural myopathies and dysmorphic findings in patients with NMD present a seriously challenging and increasing awareness of anesthesia-related complications. The mortality from anesthesia-induced MH decreased to less than 5% [4].
We performed an experience with anesthetic management of a 28-month-old preterm male patient with KDS who had an epilepsia and congenital laryngomalacia and underwent tracheostomy with a successful outcome.
The standard was written, and verbal ethical consent was obtained from the patient's legal guardian to record the information in our case report.
Case Presentation
A 28-month-old preterm male infant (weight 10 kg, height 120 cm) was born with cesarean section after the 35-weeks gestation, and his birth weight was 2580 g. His APGAR score was 6/8, with microcephaly and congenital laryngomalacia. After the delivery, the baby had a syndromic facial appearance and inadequate spontaneous respiration, he was followed up in the newborn service, and positive pressure ventilation was done. On physical examination, he had a dysmorphic face, micrognathia, prominent ears, hypertelorism, low-slanting palpebral fissures, and microcephaly. There was bilateral hearing loss.
He was referred from a peripheral hospital to scheduled open tracheostomy under general anesthesia. He was on antibiotic therapy because of recurrent airway tract infections and adjuvant respiratory therapy (nebulization and on high-flow nasal oxygenation (HFNO)) due to existing chronic respiratory failure secondary to myopathies and pectus carinatum. The child was known to have KDS. A motor milestone was delayed, and he could not stand or sit up. Additionally, the child had generalized seizures for which he was under antiepileptic therapy. No medical history of anesthetic complications or NMD increased this patient's susceptibility to malignant hyperthermia, and no family history of death from MHS.
During the preoperative evaluation, he had dysmorphic expressions characterized by the KDS (Figure 1). The cardiac examination results were normal. The neurological inspection showed diffuse muscular hypotonia and hypoactive deep tendon reflexes. On laboratory assay, his creatinine level was 0.44 mg/dL, urea 22 mg/dL, and hemoglobin 8 g/dL. The metabolic studies were normal.
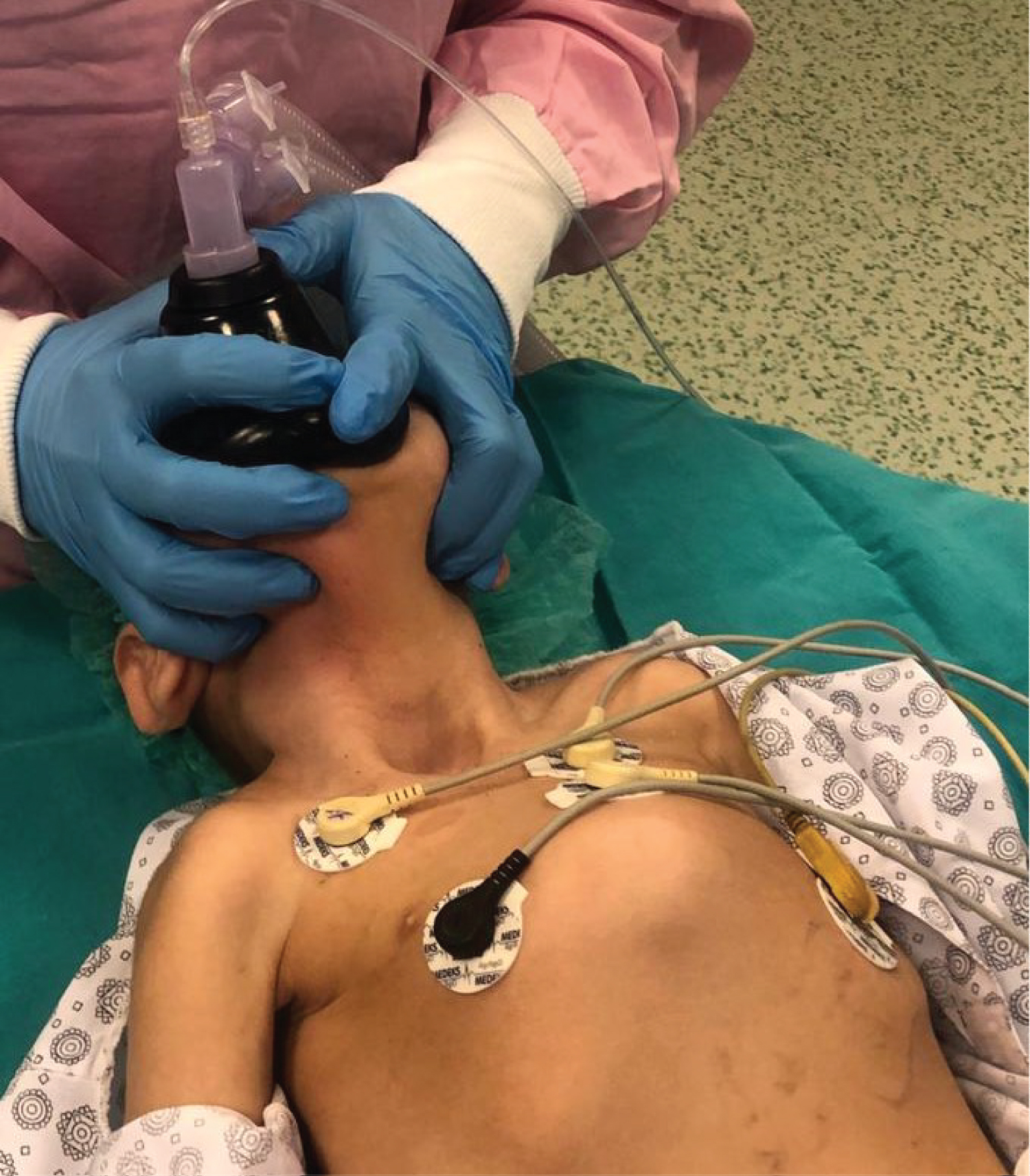
Figure 1: The patient has a dysmorphic face, micrognathia, prominent ears, hypertelorism, low-slanting palpebral fissures, microcephaly, and pectus carinatum.
The child was admitted to the operating room with complete care. The anesthetic machine was flushed with 100% oxygen at 8-10 L/min for at least 30 minutes, and the soda lime was changed. Difficult airway equipment was ready, including laryngeal mask airway (LMA), bougie, endotracheal tube (ETT) guide, video laryngoscopy, and fiber optic bronchoscopy. The otolaryngologist (ENT) surgeons were also prepared for a difficult airway to access the front-of-neck airway (FONA). There was an MH emergency kit. A bubble trap was connected to an established i.v. cannula. Standard monitoring was attached (electrocardiography, pulse oximetry, non-invasive blood pressure, temperature, and end-tidal carbon dioxide (EtCO2)) and Bispectral Index (BIS) monitoring. We used propofol 2-3 mg/kg i.v. bolus for anesthesia induction, and maintenance was done with infusions of remifentanil at 0.05-0.2 µ/kg/min and propofol at 5-10 mg/kg/hr were then commenced after the trachea was intubated successfully with a 4.0 ID endotracheal tube. No neuromuscular blocking agents were used. After ten minutes, the propofol infusion was reduced to 8 mg/kg/hour and 5 mg/kg/hour after 10 minutes. During surgery, the child's SpO2 was monitored at 90-94%, and EtCO2 remained normal, BIS 40-60, with no hemodynamic and temperature changes. The patient was warmed by an underbody blanket so that he was prevented from hypothermia. The ENT doctors opened a tracheostomy and inserted the tracheal cannula. Simultaneously, he was extubated and connected to mechanical ventilation (Figure 2). At the end of the surgery, the infusions of remifentanil and propofol were discontinued. Surgery proceeded uneventfully, and anesthesia lasted 45 minutes without any complications.
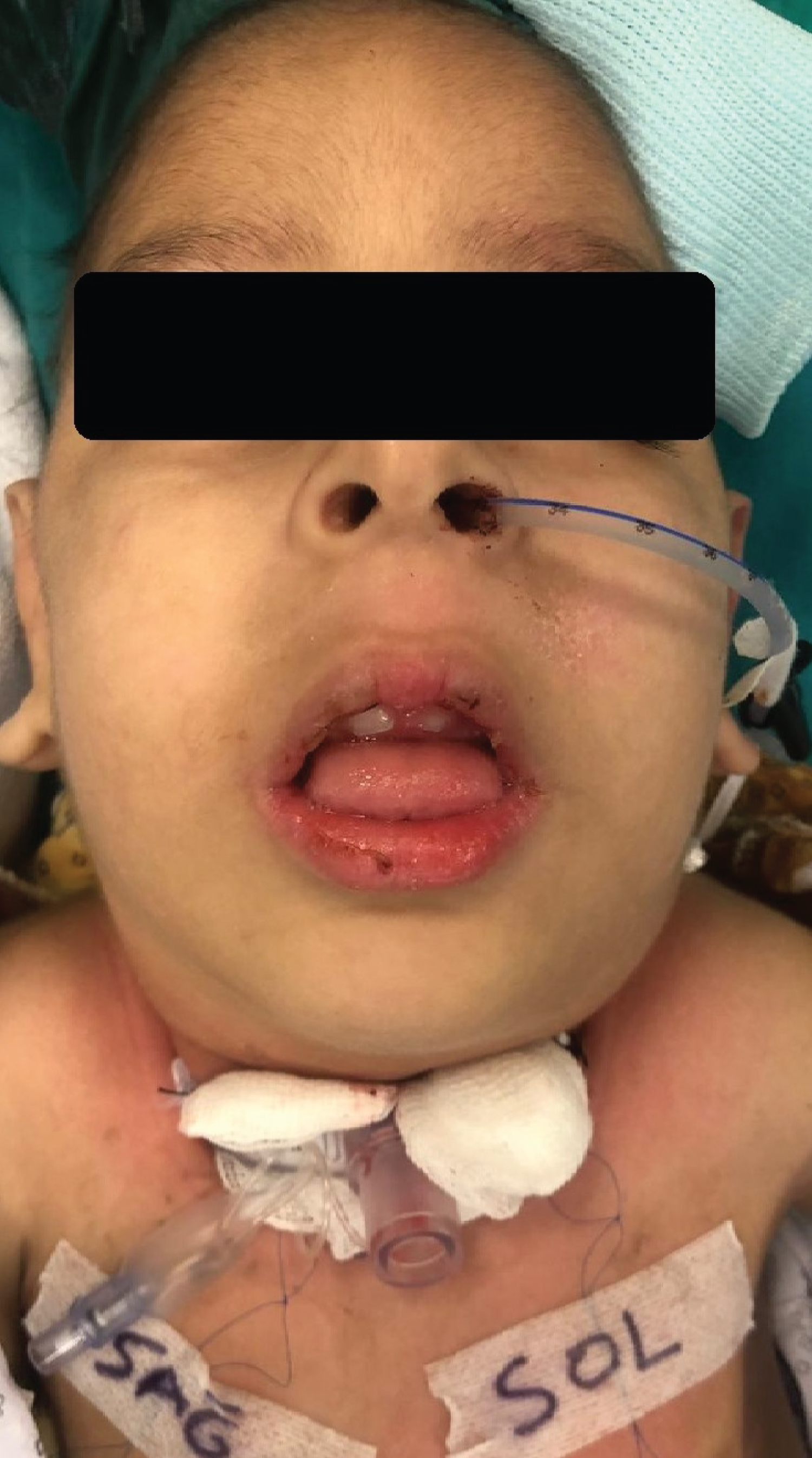
Figure 2: Tracheostomy opened, and the patient extubated.
Postoperatively, a patient was transferred to the pediatric intensive care unit (ICU), requiring ventilatory support and further observation. In the pediatric ICU, he remained stable with satisfactory arterial blood gasses (ABG) and normal temperature. The child had an unremarkable postoperative course, and he was discharged from the hospital ten days later.
Discussion
This paper report presents a successful result after tracheostomy in a patient with KDS and congenital laryngomalacia. King and Denbourogh first documented this rare syndrome in 1973 in retrospective studies of 18 males with MH [4]. Several reported cases of KDS have been ascertained, ensuing an episode of MH. In addition, Graham, et al. proposed that KDS performs a typical phenotype that may result from several different slowly progressive congenital structural myopathies [5]. These authors suppose there is some phenotypic overlap with Noonan syndrome. Our patient also had facial dysmorphic features, short stature, pectus carinatum, and structural myopathies such as congenital laryngomalacia but no cryptorchidism and low-set ears.
Identifying KDS is challenging, and there are no strict diagnostic criteria. Besides the physical manifestations, the serum creatinine phosphokinase may be elevated or normal, but it does not carry a predictive value [6]. Several studies suggest size fiber variability and type I fiber atrophy or predominance [2].
Mutations of the RYR-1 gene in the musculoskeletal structure are one of the abnormalities in patients with MHS [7]. Due to the variability of phenotype expression in patients with MHS and the confirmation of diagnosis by genetic testing of MHS is limited by expensiveness, time-consuming, not feasible, and has a low sensibility (25%) [8]. In our patient, RYR-1 receptor gene testing was not found.
The airway treatment of congenital laryngomalacia in pediatric patients is essential. Our patient had congenital laryngomalacia and myopathy, which resulted in several episodes of respiratory failure. Therefore, the ENT surgeons chose open tracheostomy over supraglottoplasty.
The main focus on perioperative management in patients with KDS is awareness of anesthesia-related complications such as MHS and potential airway problems. In most cases, the risk of an MH crisis is low, and the possibility of anesthesia-induced episodes can be decreased to zero. We decided not to use Dantrolene prophylaxis. The drug was available for emergencies in the operating room. It is recommended to starta prophylactic regimen 1 or 2 days before surgery at doses of 4-8 mg/kg-1/day [9]. Based on several studies, preventive therapy is no longer recommended in patients with MHS undergoing general anesthesia, not least because of dantrolene's adverse effects [10].
The European Malignant Hyperthermia Group (EMHG) arranged a protocol for the anesthetic workstation [11]. In our operating theater, we had a Drager Primus. Preparing this anesthetic machine for MHS patients requires flushing 100% of oxygen for 70 minutes to clear up the Anesthetic machine from inhalation agents. FGF should be maintained during anesthesia in this anesthetic machine because of a rebound increase in anesthetic concentration in the FGF [12]. We flashed at least 30 minutes with 10 L/min FGF and changed a soda-lime, anesthetic breathing circuit.
Our affected child had facial anomalies making tracheal intubation potentially difficult. The equipment for difficult airways in pediatric patients was readily feasible. LMA, bougie, ETT guide, video laryngoscopy, and fiber optic bronchoscopy were available in the operating room. Some techniques have been represented for intubation without neuromuscular blocker agents. We decided to use the propofol 2-3 mg/kg i.v. for tracheal intubation, considering his greater jaw relaxation and increased effectiveness of attenuating the laryngeal reflexes [13]. No other anesthetic agents are needed to potentiate propofol's effect regardless of myopathy's existence in our child.
Remifentanil is a potent analgesic with a short half-life promoting the quick return of respiratory and central neurological function. Elimination is independent of organ function and may be ideal for children with immature hepatic and renal functions. The pharmacokinetics of remifentanil emerges to be the same in children and adults [14]. Remifentanil is a unique drug that has a valuable role in anesthesia for children.
The hemodynamics, temperature, and EtCO2 remained normal in the intraoperative period. Since the tracheostomy opened and inserted the tracheal cannula, any potential problems with extubating and the airway were eliminated.
Postoperatively, during a 1-hour stay in the pediatric ICU, pyrexia and other hypermetabolism features such as tachypnoea, tachycardia, and hypercarbia were absent.
Anesthesia management of patients with an increased risk of developing malignant hyperthermia is essential. Patients must not be exposed to potent inhalational anesthetics or succinylcholine. To avoid these agents is to avoid general anesthesia by substituting a regional anesthetic technique if appropriate. Strategies to prevent the triggering agents are essential if general anesthesia is required. Total intravenous anesthesia could be the best choice for these patients. To clear up inhalational agents of the anesthetic machine by flushing the machine with 100% oxygen, changing soda-lime, and having a ready MH emergency kit [15].
Conclusions
We describe the outstanding management of a pediatric patient with KDS and MHS under general anesthesia. The key to successful management is a cooperative multidisciplinary team approach, and we, anesthesiologists, must be aware of and adjust anesthetic techniques.
Declaration of Interest
None.
Authors' Contribution
ON acquired the data and figures and produced the first draft of this report. AU analyzed the figures. ON and DK contributed to receivingdata, writing the manuscript, interpreting, discussing, and revising the final manuscript. NÇ and ZK helped with the conception and design, interpretation, draft writing, final manuscript editing, and approval of the final submitted manuscript.
Availability of Data and Material
The data generated or analyzed during this study are included in this article or, if absent, are available from the corresponding author upon reasonable request.
Acknowledgment
None.
Ethical Statement
Başkent University Hospital human research ethics committee approval was obtained for writing this case report.
References
- Maharaj R, Osborne I. The King-Denborough syndrome in the pediatric patient. Southern African Journal of Anaesthesia and Analgesia. 2007;13(2): 27-30.
- Reed, Umbertina Conti et al. King-Denborough Syndrome: Report of two Brazilian cases. Arquivos de Neuro-Psiquiatria. 2002;60(3-B):739-741.
- Denborough M. Malignant hyperthermia. Lancet. 1998;352(9134):1131-1136.
- King JO, Denborough MA, Zapf PW. Inheritance of malignant hyperpyrexia. Lancet 1972;1:365-370.
- Graham GE, Silver K, Arlet V, Der Kaloustian VM. King syndrome: Further clinical variability and review of the literature. Am J Med Genet. 1998;78(3):254-259.
- Isaacs H, Barlow MB. The genetic background to malignant hyperpyrexia revealed by serum creatinine phosphokinase estimations in asymptomatic relatives. British Journal Anaesthesia. 1970;42:1077-1084.
- Ronald SL, Sarah MG, James JD, Sheila Riazi; Malignant hyperthermia susceptibility and related diseases. Anesthesiology. 2018;128:159-167.
- Litman RS, Rosenberg H. Malignant hyperthermia: Update on susceptibility testing. JAMA. 2005;293(23):2918-2924.
- Britt BA. Dantrolene. Canadian Anesthetists' Society Journal. 1984;31:61-75.
- Krause T, Gerbershagen MU, Fiege M, Weithorn R, Wappler F. Dantrolene - A review of its pharmacology, therapeutic use, and new developments. Anaesthesia. 2004;59:364-373.
- Henrik R, Börge B, Diana B, Thierry G, Sebastian H. et al.Consensus guidelines on perioperative management of malignant hyperthermia suspected or susceptible patients from the European Malignant Hyperthermia Group. British Journal of Anaesthesia. 2021;126(1):120-130.
- Prinzhausen H, Crawford MW, O'Rourke J, Petroz GC. Preparation of the Dräger Primus anesthetic machine for malignant hyperthermia-susceptible patients. Can J Anaesth. 2006;53(9):885-890.
- Shaikh SI, Bellagali VP. Tracheal intubation without neuromuscular block in children. Indian J Anaesth. 2010;54(1):29-34.
- Ross AK, Davies PJ, Dear Gdel, et al. Pharmacokinetics of remifentanil in pediatric patients undergoing elective surgery or diagnostic procedures. Anesth Anal. 2001;93:1393-1401.
- Hopkins PM, Girard T, Dalay S, Jenkins B, Thacker A, Patteril M, et al. Malignant hyperthermia: Guideline from the Association of Anaesthetists. Anaesthesia. 2021;76(5):655-664.