Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/188
Review Article | Volume 11 | Issue 2 Open Access
Interleukin-33: A Double-Edged Sword in Sepsis
Shuai Liu1, Yinyan Yue1, Qiuge Wu1* and Li-Ming Zhang2*
1Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
2Department of Anesthesiology & Perioperative Medicine, University of Pittsburgh School of Medicine and University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA
Dr. Qiuge Wu, The First Affiliated Hospital of Zhengzhou University, Erqi District, Zhengzhou 450052, Henan Province, China, Tel: (+86)-13526813392, Fax: (+86)-37166913346, E-mail: fccwuqg@zzu.edu.cn;Dr. Li-Ming Zhang, Department of Anesthesiology & Perioperative Medicine, UPMC, 200 Lothrop Street, Pittsburgh, Pennsylvania 15213, USA, Tel: 412-648-6077, Fax: 412-648-6014, E-mail: zhangl1@anes.upmc.edu
Editor: Renyu Liu, MD; PhD; Professor, Department of Anesthesiology and Critical Care, Perelman School of Medicine at the University of Pennsylvania, Center of Penn Global Health Scholar, 336 John Morgan building, 3620 Hamilton Walk, Philadelphia, PA 19104, USA, Fax: 2153495078, E-mail: RenYu.Liu@pennmedicine.upenn.edu
Received: September 12, 2024 | Accepted: September 13, 2024 | Published: September 16, 2024
Citation: Liu S, Yue Y, Wu Q, Li-Ming Z. Interleukin-33: A Double-Edged Sword in Sepsis. Transl Perioper Pain Med 2024; 11(2):612-619
Abstract
Despite some advances that have been made in the clinical management of sepsis, its morbidity and mortality rates remain high. The pathogenesis of systemic inflammation and organ damage leading to septic mortality is not fully understood. Interleukin-33 (IL-33), a new member of the IL-1 superfamily, can be a traditional cytokine and a nuclear factor regulating gene transcription. Due to its biological characteristics, IL-33 can act as a double-edged sword in sepsis: It not only contributes to clear bacteria and improves survival but also causes inflammation, immunosuppression, and organ damage via modulating immune response. In this review, we will summarize updated information on the dual functions of IL-33 in the host immune response to sepsis.
Keywords
Interleukin-33 (IL-33), Pro-inflammatory, Anti-inflammatory, Sepsis
Introduction
In 2005, Interleukin-33 (IL-33) was identified as a new member of the interleukin (IL)-1 cytokine family [1]. IL-33 is constitutively and abundantly expressed in bronchial and intestinal epithelial cells, endothelial cells, and fibroblasts in human [2,3]. Under resting conditions, IL-33 is localized in the nucleus, as mediated by the presence of an N-terminal domain nuclear localization sequence. Since IL-33 can function both as an intracellular nuclear factor and a conventional cytokine, which makes it a particular characteristic role in transcriptional regulation and activation of the ST2 receptor complex signal. IL-33 has been proposed to be another alarmin with dual functions, similar to HMGB1 and IL-1α. Accumulated experimental evidence indicates that IL-33 is involved in both Th1- and Th2-cells mediated immunity responses and has both pro-inflammatory and anti-inflammatory effects simultaneously [4].
Sepsis remains a leading cause of death in the Intensive Care Unit (ICU) [5]. There is increasing evidence that suggests that IL-33 is involved in the initiation and progression of inflammatory diseases, such as sepsis [6,7]. However, controversy remains with its definitive role in sepsis. In this review, we provide recent advances in IL-33-mediated immune regulation and its effect on sepsis.
Molecular Features of IL-33
The location of IL33 gene has been mapped to chromosome 9p24.1 in humans, while its mouse homolog is found on the syntenic chromosome 19qC1 region [1]. The IL-33 protein is composed of three functional domains: Nuclear domain, central domain, and IL-1-like cytokine domain [8]. Under basal conditions, the N-terminus of IL-33 contains an evolutionarily conserved homeodomain-like helix-turn-helix (HTH) DNA binding domain, which is necessary and sufficient for nuclear targeting and is involved in the repression of transcription that has been ascribed to IL‐33 [9]. The central domain of IL-33 is sensitive to neutrophil- and mast cell-derived proteases due to the protease cleavage sites in it [10,11]. As to the C-terminal IL-1-like cytokine domain of IL-33, its binding interface structure plays a critical role in interacting with all three Ig-like domains of the ST2 ectodomain [12], and mediates the cytokine activities [13] (Figure 1). Consistent with the features in structure, thus, IL-33 might have dual functions involved in immune regulation either as a traditional cytokine through activation of the ST2 receptor complex or as an intracellular nuclear factor with transcriptional regulatory properties, similar functions have previously been known for IL-1a and HMBG1 [14].
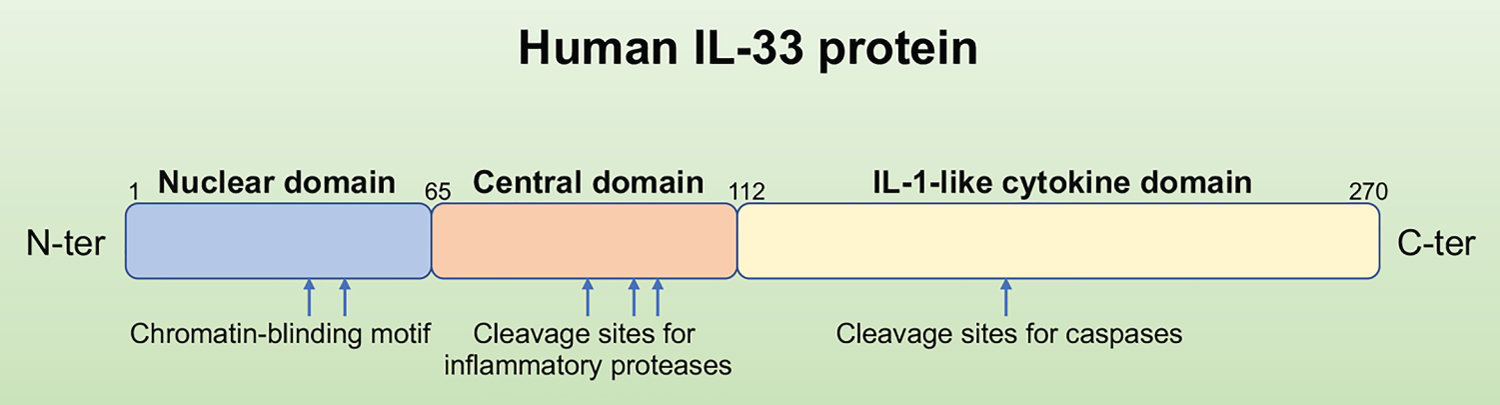
Figure 1: Primary structure of the human IL-33 protein. The full-length IL-33 is a protein with 270 amino acids. It has the N-terminal nuclear structural domain and the C-terminal IL-1-like cytokine structural domain, the cleavage sites for proteases involved in IL-33 activation (inflammatory proteases) or inactivation (caspases) are shown above.
Expression of IL-33
During homeostasis, constitutive and abundant expression of IL-33 mRNA and protein is found in the airway, bronchial epithelial cells, endothelial cells of high endothelial venules, fibroblasts, and platelets in human [1,2,15,16]. In contrast, in rodents, constitutive expression of IL-33 is not detected in endothelial cells under basal conditions, however, its expression can be induced in the microenvironment of chronic inflammation, revealing the species-specific differences between human and mouse [3]. Pichery, et al. found that endogenous IL-33 is highly expressed in mouse epithelial barrier tissues (including stratified squamous epithelia from the vagina and skin, cuboidal epithelium from the lung, stomach, and salivary gland), lymphoid organs, brain, embryos during homeostasis [3]. Under the condition of inflammation or tissue stress, the constitutive expression of IL-33 at a high level in tissues can be further upregulated, such as in the lung during papain-induced allergic airway inflammation, and in the liver during LPS-induced endotoxin shock [3].
Processing and Release of IL-33
While in nuclear, IL-33 has a crucial role in chromatin compaction and repression of gene expression [17]. In addition to repressing gene expression, IL-33 nuclear location was demonstrated to have an ability to potently regulate the extracellular function of IL-33; in the absence of nuclear localization sequestration, IL-33 is sustainably released into the extracellular from the nucleus, which ultimately causes non-resolving lethal inflammation, suggesting that the sub-cellular localization of IL-33 is very important for controlling its extracellular release and maintaining immune homeostasis [18]. Furthermore, because the release of IL-33 is independent of caspase-1, caspase-8, or calpain [19], the nuclear localization of IL-33 becomes even more important: after release, the full-length form of IL-33 is active and may contribute to the pathogenesis of inflammatory response in many diseases. The activity of the full-length form of IL-33 is abolished when processed by caspase-1, caspase-3, or caspase-7 [20,21]. Once the full-length form of IL-33 (30 kDa) is released into the extracellular space, it is cleaved by proteases, such as cathepsin G, calpain, or neutrophil elastase, then leads to the production of C-terminal cleavage products containing the entire IL-1-like domain, finally becomes to a stronger pro-inflammatory form, mature IL-33 (18 kDa) [11] (Figure 2).
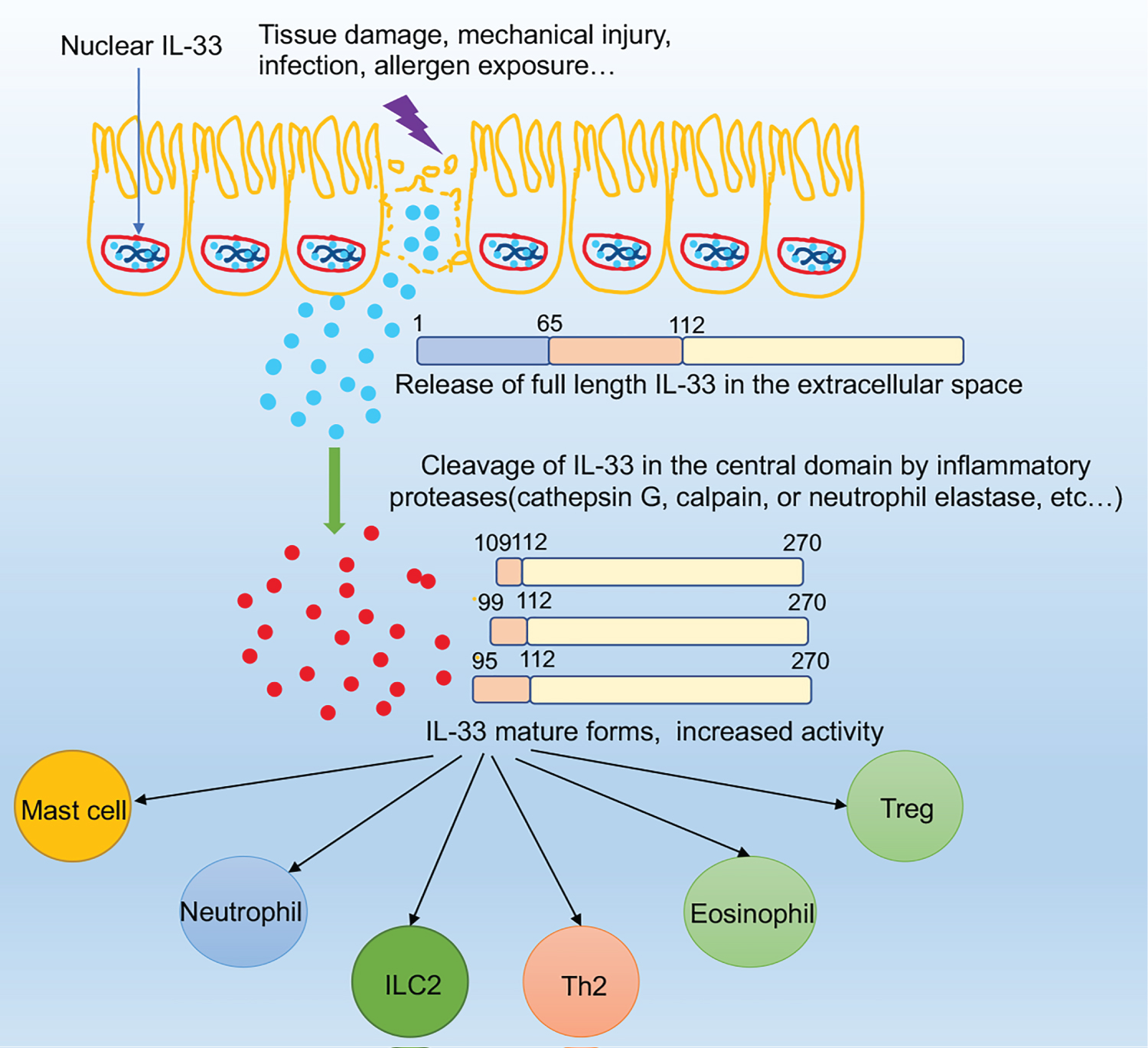
Figure 2: The mechanisms of IL-33 extracellular release. After epithelial cell exposure to tissue damage, mechanical injury, infection, allergen exposure, etc. IL-33 will be quickly released from the nucleus and active the ST2+ cells such as ILC2s. In this process, IL-33 can be cleaved into shorter mature forms by inflammatory proteases such as neutrophils cathepsin G.
Pichery, et al. found that the IL-33 protein was only localized in the cell nucleus rather than in the cytoplasmic of producing cells [3]. However, Kakkar and her colleagues demonstrated that IL-33 localized simultaneously to nuclear euchromatin and cytoplasmic vesicles, newly synthesized IL-33 molecules are initially imported into the nucleus for euchromatin association, from where they gradually efflux into the cytoplasmic space to be packaged into secretory vesicles during biomechanical overload by living cells, which was dependent on nuclear pore complex function [22].
After being released into the extracellular space, the oxidation of IL-33 occurs rapidly by the formation of two disulfide bridges, which leads to the biological activity of IL-33 at its receptor ST2 being terminated [23]. Such a mechanism would limit IL-33 in time and space, restricting its activity to the ST2-dependent immunological responses. This suggests that the activity of IL-33 in mediating inflammatory response is also regulated by post-translational modification.
IL-33/ST2 Signaling
Once released, IL-33 can induce signaling pathways through binding to its heterodimeric receptor complex which is composed of ST2 and IL-1R accessory protein (IL-1RAP) [24]. ST2, the receptor of IL-33, is a member of the IL-1 receptor (IL-1R) family. The two major protein isoforms encoded by the ST2 gene include a transmembrane full-length form (ST2 or ST2L) and a soluble, secreted form (sST2) [6,25]. Membrane-bound ST2 is thought to be a functional component for IL-33 signaling [6,25], whereas sST2 acts as a decoy receptor for blocking IL-33 signaling due to a lack of transmembrane and intracellular domains [26].
Upon activation, IL-33/ST2/IL-1RAP leads to the recruitment of myeloid differentiation primary response protein 88 (MyD88) [27], interleukin receptor-associated kinase 4 (IRAK4), followed by interaction between IRAK1, IRAK2, and/or IRAK3, together forms the complex called myddosome. This myddosome then interacts with tumor necrosis factor receptor-associated factor 6 (TRAF6) [28,29], leading to the activation of the transcription factors, including nuclear factor-kappaB (NF-κB) and MAP kinases (ERK, p38 and JNK) [1,6,24].
Subsequently, IL-33 induces the production of various pro- or anti-inflammatory mediators such as IL-6, TNF-α, IL-1β, IL-5, and IL-13 in Th2 cells, eosinophils, basophils, and mast cells, et al. [27,30-32]. Furthermore, IL-33 was also involved in driving both Th1 and Th2 immune responses through its activity on a variety of immune cells under specific microenvironments, which eventually results in a complex range of biological functions in different diseases [6,7] (Figure 3).
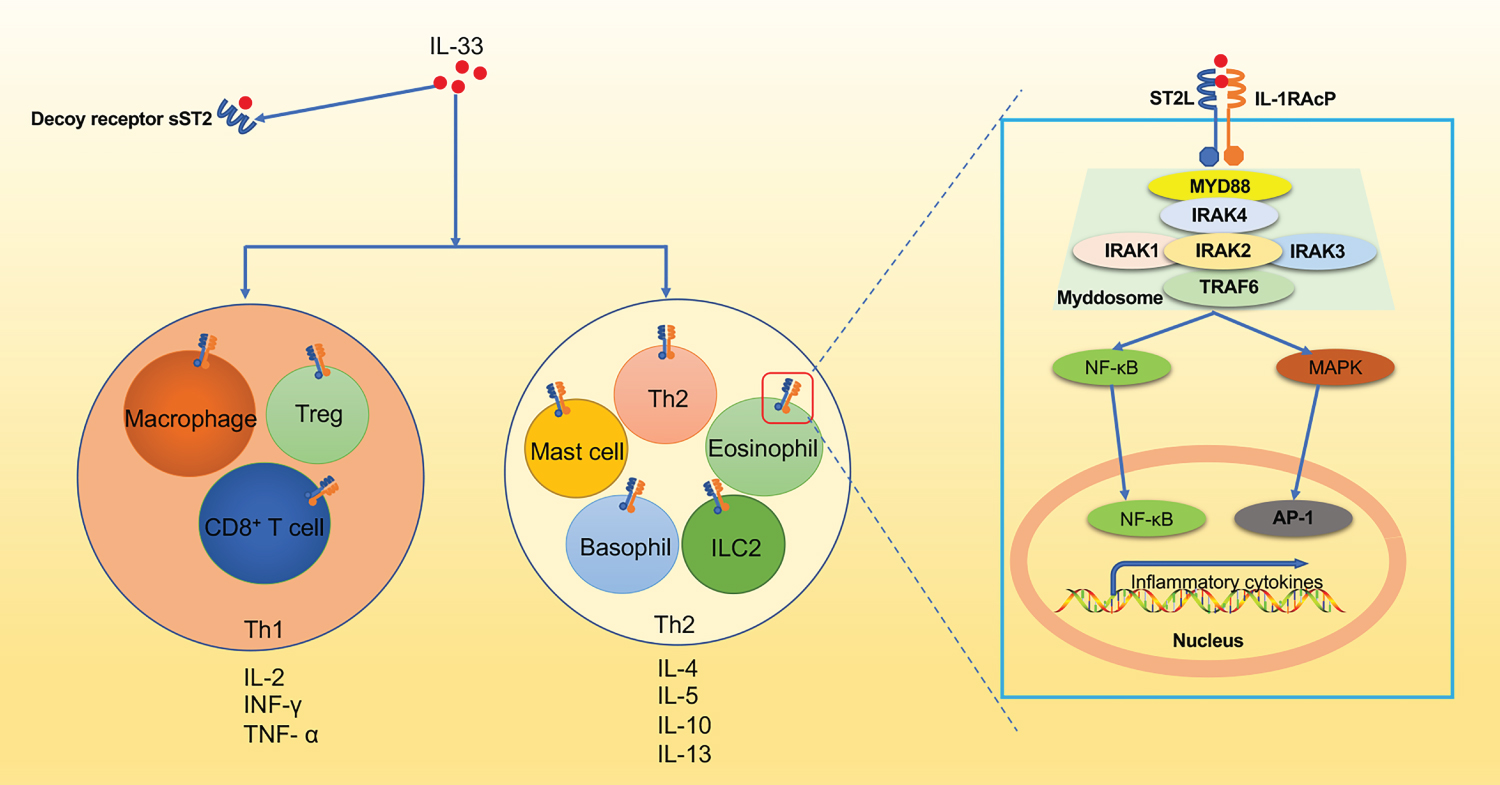
Figure 3: The IL-33-ST2 signaling pathway. IL-33 activates ST2+ Treg, CD8+ T cells, and macrophages to produce TNF-α and IFN-γ to mediate type I immune responses. Additionally, ST2+ mast cells, basophils, eosinophils, ILC2s, and Th2 cells are also activated by IL-33 to produce type 2 cytokines (IL-4, IL-5, IL-9, IL-13) promoting the classical type 2 immune response. At the molecular level, IL-33 forms a complex with ST2 and IL-1 receptor accessory protein (IL-1RAcP). Subsequently, myeloid differentiation primary response protein 88 (MYD88), IRAK4 is then recruited to MyD88, followed by interaction between IRAK1, IRAK2, and/or IRAK3 to form a complex known as the myddosome. This myddosome then interacts with TRAF6 and further activates the transcription factors NF-κB or MAPK, which lead to NF-κB and AP-1 pathways respectively.
The Role of IL-33 in Sepsis
Clinical data have shown elevated serum levels of IL-33 or sST2 in adult patients with sepsis, whereas more serum sST2 levels were present in the non-survivors compared to the survivors [33-36]. There are significantly higher levels of IL-33 or sST2 in blood in the early phase of childhood sepsis [37,38]. Ziehe’s results showed that the increase in IL-33 concentration was positively correlated with the expression of aquaporin-3 (AQP3), accompanied by a shortened survival period in patients with sepsis [39]. Even though some scholars have concluded that sST2 level is a valuable marker in assessing the severity and mortality of sepsis [40-42], even identifying the etiology of shock between septic shock and cardiogenic shock in the early phases [43]. Additionally, elevated levels of ST2 are associated with a higher risk of hyperglycemia in mechanically ventilated septic patients [44]. However, further research is needed to elucidate whether IL-33 can be used as a biomarker to diagnose, assess the severity, and predict the outcome of sepsis.
Furthermore, some animal experimental studies showed that IL-33 plays a protective role against sepsis. In a sepsis model induced by cecal ligation and puncture (CLP), the administration of exogenous IL-33 enhances bacterial clearance, leading to improved survival of septic mice; at 24 h after CLP, IL-33 attenuates the system inflammation, represented by the decreased levels of IL-6, IL-10, TNF-α, and IFN-γ in serum, as well as the severity of organ injury, which was likely due to IL-33 prevented apoptosis of T lymphocytes and improved bacterial clearance [45]. Recombinant IL-33 protects against LPS-induced endotoxemia and liver injury by increasing the number of liver-infiltrating ST2+ Tregs and promoting the early resolution of excessive inflammation [36]. Lai, et al. found that IL-33/ST2 signaling increases the expression of GRK2 and decreases the level of CXCR4, mediating ILC2 expansion in the lungs and subsequently secretion of IL-9, which plays an important role in protecting lung endothelial cells from pyroptosis in sepsis mice [46,47]. It is also reported that IL-33 treatment suppressed pro-inflammatory cytokines production and improved the survival rate in sepsis mice by suppressing the IL-17 pathway through activating the suppressor of cytokine signaling (SOCS)-3 [34]. Moreover, exogenous IL-33 treatment can increase the neutrophil influx into the peritoneal cavity, which contributes to enhancing bacterial clearance in the site of infection and reducing mortality in mice with sepsis from CLP [48]. Interestingly, the recruitment of neutrophils influx from the circulation to the site of infection was mediated by the CXCR2, a chemokine receptor on neutrophil, which was down-regulated via activation of Toll-like receptors (TLRs) in circulating neutrophils during severe sepsis [49]. But IL-33 can reverse the down-regulation of CXCR2 expression and promote neutrophil recruitment by repressing the expression of G protein-coupled receptor kinase-2 (GRK2) [48], a serine-threonine protein kinase that induces internalization of chemokine receptors [50,51]. Similarly, increased neutrophil influx into the focus of infection caused by IL-33 contributes to reducing bacterial loads was also observed when sepsis mice infected with gram-negative bacteria or acute Staphylococcus aureus [52,53]. Most recently, Gong, et al. demonstrated that Sphingosine-1-phosphate receptor 2 (S1PR2) deficiency modulated the type 2 immune response by promoting the IL-33 increase in macrophage, accompanied by reduced lung injury in post-septic mice [54]. Taken together, an early phase of neutrophil-mediated bacterial clearance seems to be a powerful strategy for IL-33 to benefit bacterial sepsis in the early phases. However, in the late phase of uncontrolled sepsis, the downside of the sustainable enhanced neutrophil recruitment mediated by IL-33 may cause secondary remote organ damage in the late phase of sepsis. Thus, precise modulation in different stages of disease progression is very critical.
Contrary to the view mentioned above, some scholars have found that IL-33 plays a role in promoting the progress of sepsis. IL-33 was found to drive systemic inflammation, ILC2 IL-5 expression as well as neutrophil and monocyte infiltration and cytokine production in the lungs, which identifies IL-33 as a driver of systemic and pulmonary inflammation during intra-abdominal sepsis [55]. IL-33 disturbs medullary thymic epithelial cell (mTEC)/cortical TEC (cTEC) compartment, which results in immunosuppression by inducing thymic involution-associated naive T cell aging and impairs host control of severe infection in mouse disease models of schistosomiasis or sepsis [56]. IL-33-deficient mice were resistant to endotoxin shock in vivo and showed a substantially diminished LPS-induced systemic inflammatory response (IL-6, IL-1α, and IL-1β) at 9 and/or 48 h after LPS stimulation in vitro [57]. Recombinant IL-33 further enhanced the LPS-induced inflammatory cytokine releasing (IL-6, TNF-α, and IL-1β) in mouse macrophages in an ST2-dependent manner [58,59]. The reason why IL-33 was able to enhance macrophages response to LPS is being linked to IL-33 treatment triggers an increase in the expression of the LPS receptor components (MD2/CD14 and TLR-4) at the plasma membrane, the level of the soluble form of CD14 (sCD14) and the MyD88 adaptor molecule [58]. Apart from the enhanced expression of pro-inflammatory factors (e.g., iNOS, IL-6, and TNF-α) induced by LPS in bone marrow-derived macrophages (BMDMs), IL-33 was showed directly activate macrophage the expression of MHC class I, MHC class II, CD80/CD86, as well as inducible NO synthase (iNOS) in a dose-dependent manner [60]. Moreover, Nascimento’s results showed that endogenous IL-33, released during sepsis, induces the switch of macrophages toward M2 polarization, which has an essential function in the expansion of the Treg cell population via the production of IL-10 [33]; another study found that exogenous IL-33 resulted in the expansion of TIGIT+ Tregs depending on the STAT6 and M2 macrophages, contributing to the development of long-term sepsis-induced immunosuppression [61]. In addition, IL-33 facilitates the level of macrophage pyroptosis by activating the NF-κB/p38 MAPK signaling pathway and increases the mortality of mice with sepsis [62].
Our recent study indicates that in a two-hit model of CLP followed by moderate mechanical ventilation (MTV) or low tidal volume (LTV), the extent of inflammation and injury correlated with intra-pulmonary IL-33 levels and deletion of IL-33 or ST2 attenuated lung injury in the two-hit model [63]. Consistently, our in vitro experiments also found that cyclic stretch was able to induce IL-33 production through the HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells [64]. Furthermore, in our previous study, we have identified that WNT1-inducible signaling pathway protein 1 (WISP1) plays an important role in ventilator-induced lung injury (VILI) and CLP-induced ALI [65-67]. Interestingly, in our most recent study, we found that IL-33 can mediate the expression of WISP1 by activating AKT- and ERK-GSK-β-catenin signaling pathways in macrophage via the interaction of β-catenin/TCF/CBP/p300 in the nucleus, which finally contributes to the occurrence and development of lung injury in the two-hit lung injury model [68]. These findings place IL-33 as a proximal driver of lung injury induced by mechanical ventilation in intra-abdominal sepsis. Understanding the intrinsic link between IL-33, mechanical ventilation, and sepsis is fundamentally important for the development of rational strategies for the treatment of different stages of sepsis and the prevention of its associated complications.
Conclusion
Taken together, these findings suggest that IL-33 plays an essential dual-functional role in the progression of sepsis, the role of IL-33 in sepsis appears to be time and concentration-dependent (Figure 4). However, the exact function and potential mechanism of the IL-33 signaling pathway in sepsis remains to be elucidated. Further investigation of the role of IL-33 in sepsis may lead to the development of new therapeutic targets and strategies.
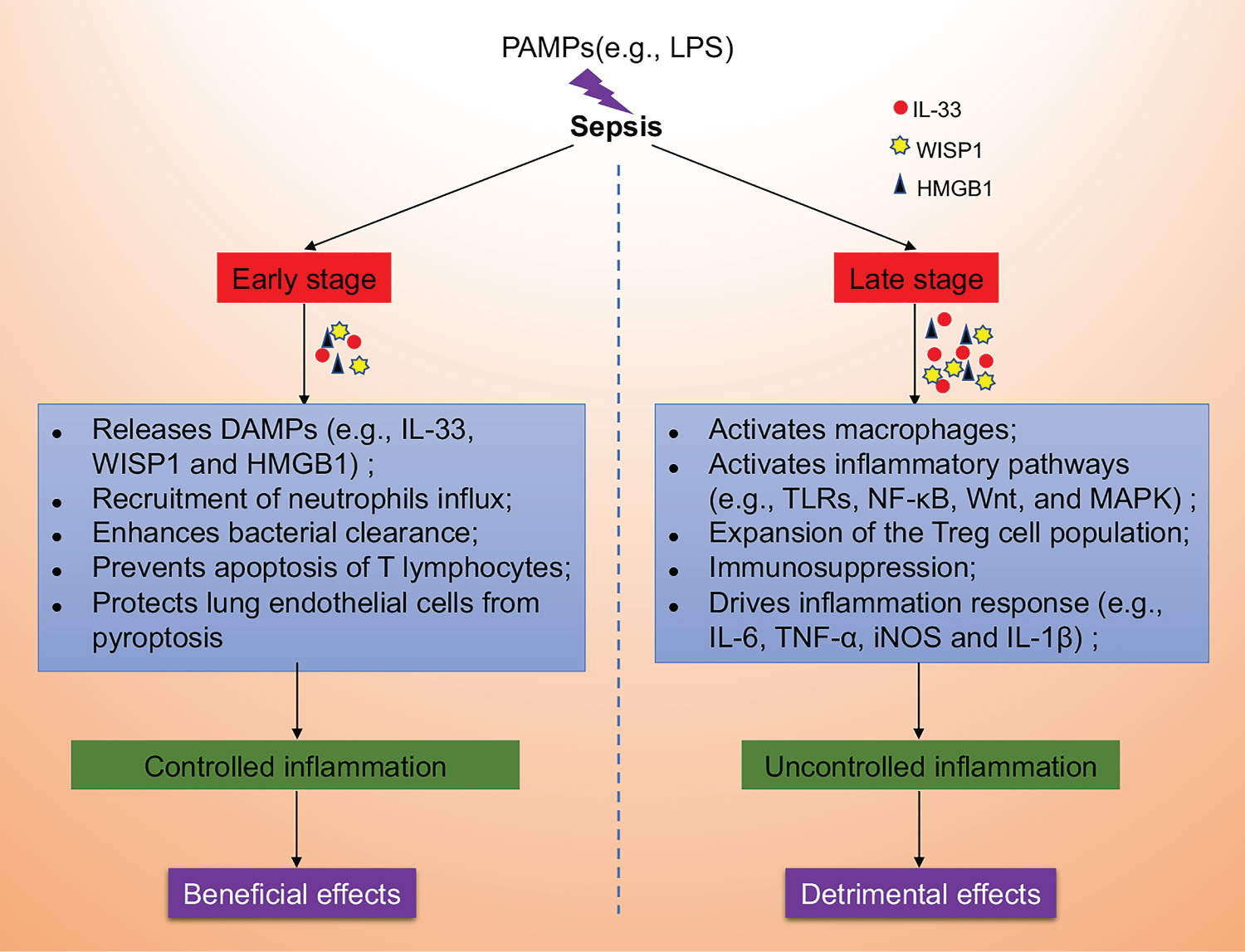
Figure 4: The dual-function of IL-33 in sepsis. We speculate that pathogen-associated molecular patterns (PAMPs, e.g., LPS) induce the release of damage-associated molecular patterns (DAMPs, e.g., IL-33, WISP1 and HMGB1) in the early stage of sepsis, which plays a beneficial role in by recruiting neutrophils influx, enhancing bacterial clearance, preventing apoptosis of T lymphocytes and protecting lung endothelial cells from pyroptosis in controlling inflammation. However, with the development of sepsis towards late stage, the sustainable and increased release of IL-33 may exert detrimental effects through enhanced and uncontrolled inflammation by activating macrophages, activating inflammatory pathways (e.g., TLRs, NF-κB, Wnt, and MAPK), expanding of the Treg cell population, inducing immunosuppression and driving inflammation response (e.g., IL-6, TNF-α, iNOS and IL-1β).
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (82100092), the Natural Science Foundation of Henan Province of China (232300420255), the Henan Province Medical Science and Technology Research Plan (LHGJ20200380 and LHGJ20200341) and the National Institute of Health grant (R01-GM-108639).
Conflict Interests Disclosure
The authors have no conflicting interests to disclose.
References
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, et al. (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23: 479-490.
- Moussion C, Ortega N, Girard JP (2008) The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel 'alarmin'?. PLoS One 3(10): e3331.
- Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A. (2012) Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: In situ analysis using a novel Il-33-LacZ gene trap reporter strain. Journal of Immunology 188(7): 3488-3495.
- Liew FY, Pitman NI, Mcinnes IB (2010) Disease-associated functions of IL-33: The new kid in the IL-1 family. Nature Reviews Immunology 10(2): 103.
- Lakshmikanth Lakshminarayana C, Jacob Petsel S, Marathe Kedihithlu G, Chaithra Hanumanthareddy V, Castro-Faria-Neto CD Hugo (2016) Sepsis: In search of cure. Inflammation research.
- Milovanovic M, Volarevic V, Radosavljevic G, Jovanovic I, Pejnovic N, et al. (2012) IL-33/ST2 axis in inflammation and immunopathology. Immunologic Research 52(1-2): 89-99.
- Kurowska-Stolarska M, Hueber A, Stolarski B, Mcinnes IB (2011) Interleukin-33: A novel mediator with a role in distinct disease pathologies. Journal of Internal Medicine 269(1): 29-35.
- Cayrol C, Girard JP (2014) IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Current Opinion in Immunology, 31(3): 31-37.
- Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, et al. (2007) IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proceedings of the National Academy of Sciences of the United States of America 104(1): 282.
- Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, et al. (2014) Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proceedings of the National Academy of Sciences of the United States of America 111(43): 15502.
- Lefrançais E, Roga S, Gautier V, Gonzalez-De-Peredo A, Monsarrat B, et al. (2012) IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proceedings of the National Academy of Sciences of the United States of America 109(5): 1673-1678.
- Liu X, Hammel M, He Y, Tainer JA, Jeng US, et al. (2013) Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA 110(37): 14918-14923.
- Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, et al. (2009) Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors--insight into heterotrimeric IL-1 signaling complexes. Structure 17(10): 1398-1410.
- Gadina M, Jefferies CA (2007) IL-33: A sheep in wolf's clothing?. Sciences Stke Signal Transduction Knowledge Environment 2007(390): pe31.
- Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, et al. (2010) Increased IL-33 expression by epithelial cells in bronchial asthma. Journal of Allergy & Clinical Immunology 125(3): 752-754.
- Takeda T, Unno H, Morita H, Futamura K, Emisugie M, et al. (2016) Platelets constitutively express IL-33 protein and modulate eosinophilic airway inflammation. Journal of Allergy & Clinical Immunology 138(5): 1395-1403.e1396.
- Roussel L, Erard M, Cayrol C, Girard JP (2008) Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. Embo Reports 9(10): 1006-1012.
- Bessa J, Meyer CA, Mc DVM, Schlicht S, Smith SH, et al. (2014) Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. Journal of Autoimmunity 55: 33-41.
- Ohno T, Oboki K, Kajiwara N, Morii E, Aozasa K, et al. (2009) Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. Journal of Immunology 183(12): 7890-7897.
- Cayrol C, Girard JP (2009) The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proceedings of the National Academy of Sciences of the United States of America 106(22): 9021.
- Lüthi AU, Cullen SP, Mcneela EA, Duriez PJ, Afonina IS, et al. (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31(1): 84-98.
- Kakkar R, Hei H, Dobner S, Lee RT (2012) Interleukin 33 as a mechanically responsive cytokine secreted by living cells. Journal of Biological Chemistry 287(9): 6941.
- Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, et al. (2015) Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nature Communications 6: 8327.
- Chang J, Xia YF, Zhang MZ, Zhang LM (2016) IL-33 Signaling in Lung Injury. Transl Perioper Pain Med 1(2): 24-32.
- Oboki K, Nakae S, Matsumoto K, Saito H (2011) IL-33 and airway inflammation. Allergy Asthma & Immunology Research 3(2): 81-88.
- Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y (2002) Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clinical & Experimental Allergy 32(10): 1520-1526.
- Cohen P (2014) The TLR and IL-1 signalling network at a glance. Journal of Cell Science 127(11): 2383-2390.
- Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, et al. (2009) An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. Journal of Biological Chemistry 284(37): 25404-25411.
- Lin SC, Yu-Chih L, Hao W (2010) Helical assembly in the MyD88:IRAK4:IRAK2 complex in TLR/IL-1R signaling. Nature 465(7300): 885.
- Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, et al. (2007) IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proceedings of the National Academy of Sciences of the United States of America 104(47): 18660-18665.
- Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H (2008) A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol 121(6): 1484-1490.
- Kurowskastolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, et al. (2009) IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. Journal of Immunology 183(10): 6469-6477.
- Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, et al. (2017) IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nature Communications 8: 14919.
- Lv R, Zhao J, Lei M, Xiao D, Yu Y, et al. (2017) IL-33 Attenuates Sepsis by Inhibiting IL-17 Receptor Signaling through Upregulation of SOCS3. Cellular Physiology & Biochemistry 42(5): 1961-1972.
- Chun TT, Chung CS, Fallon EA, Hutchins NA, Clarke E, et al. (2018) Group 2 Innate Lymphoid Cells (ILC2s) Are Key Mediators of the Inflammatory Response in Polymicrobial Sepsis. Am J Pathol 188(9): 2097-2108.
- Wang P, Shi B, Wang C, Wang Y, Que W, et al. (2022) Hepatic pannexin-1 mediates ST2(+) regulatory T cells promoting resolution of inflammation in lipopolysaccharide-induced endotoxemia. Clin Transl Med 12(5): e849.
- F Ç, Fidanci MK, Ayar G, Saldir M, Karaoglu A, et al. (2016) Diagnostic Value of Upar, IL-33, and ST2 Levels in Childhood Sepsis. Clinical Laboratory 62(5): 751-755.
- Yang KD, He Y, Xiao S, Ai Q, Yu JL (2020) Identification of progranulin as a novel diagnostic biomarker for early-onset sepsis in neonates. Eur J Clin Microbiol Infect Dis 39(12): 2405-2414.
- Ziehe D, Marko B, Thon P, Rahmel T, Palmowski L, et al. (2024) The Aquaporin 3 Polymorphism (rs17553719) Is Associated with Sepsis Survival and Correlated with IL-33 Secretion. Int J Mol Sci 25(3).
- Hoogerwerf JJ, Tanck MWT, Van ZMAD, Xavier W, Pierre-François L, et al. (2010) Soluble ST2 plasma concentrations predict mortality in severe sepsis. Intensive Care Med 36(4): 630-637.
- Mina H, Hanah K, Hyunjeong K, Yang HS, Magrini L, et al. (2015) Soluble ST2 has a prognostic role in patients with suspected sepsis. Annals of Laboratory Medicine 35(6): 570-577.
- Eric S, Zaric RZ, Jevdjic J, Drakulic SM, Stanojevic I, et al. (2023) Interleukin 33, soluble suppression of tumorigenicity 2, interleukin 27, and galectin 3 as predictors for outcome in patients admitted to intensive care units. Open Med (Wars) 18(1): 20230859.
- Parenica J, Malaska J, Jarkovsky J, Lipkova J, Dastych M, et al. (2012) Soluble ST2 levels in patients with cardiogenic and septic shock are not predictors of mortality. Experimental & Clinical Cardiology 17(4): 205-209.
- Farooq N, Chuan B, Mahmud H, El Khoudary SR, Nouraie SM, et al. (2021) Association of the systemic host immune response with acute hyperglycemia in mechanically ventilated septic patients. PLoS One 16(3): e0248853.
- Li S, Zhu FX, Zhao XJ, An YZ (2015) The immunoprotective activity of interleukin-33 in mouse model of cecal ligation and puncture-induced sepsis. Immunology Letters 169: 1-7.
- Lai D, Tang J, Chen L, Fan EK, Scott MJ, et al. (2018) Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis 9(3): 369.
- Lai D, Chen W, Zhang K, Scott MJ, Li Y, et al. (2022) GRK2 regulates group 2 innate lymphoid cell mobilization in sepsis. Mol Med, 28(1): 32.
- JC A-F, F S, FO S, A F, Jr VW, et al. (2010) Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nature Medicine 16(6): 708-712.
- Alvesfilho JC, Freitas A, Souto FO, Spiller F, Paulaneto H, et al. (2009) Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proceedings of the National Academy of Sciences of the United States of America, 106(10): 4018.
- Vroon A, Heijnen CJ, Kavelaars A (2006) GRKs and arrestins: Regulators of migration and inflammation. Journal of Leukocyte Biology 80(6): 1214-1221.
- Penela P, Ribas C, Jr MF (2003) Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cellular Signalling 15(11): 973-981.
- Fang L, Yuan B, Tao L, Luo X, Ping H, et al. (2016) Interleukin-33 facilitates neutrophil recruitment and bacterial clearance in S. aureus -caused peritonitis. Molecular Immunology 72: 74-80.
- Ramirez-Moral I, Blok DC, Bernink JH, Garcia-Laorden MI, Florquin S, et al. (2021) Interleukin-33 improves local immunity during Gram-negative pneumonia by a combined effect on neutrophils and inflammatory monocytes. J Pathol 253(4): 374-383.
- Gong C, Jin Y, Wang X, Mao J, Wang D, et al. (2024) Lack of S1PR2 in Macrophage Ameliorates Sepsis-associated Lung Injury through Inducing IL-33-mediated Type 2 Immunity. Am J Respir Cell Mol Biol 70(3): 215-225.
- Xu H, Xu J, Xu L, Jin S, Turnquist HR, et al. (2018) Interleukin-33 contributes to ILC2 activation and early inflammation-associated lung injury during abdominal sepsis. Immunol Cell Biol 96(9): 935-947.
- Xu L, Wei C, Chen Y, Wu Y, Shou X, et al. (2022) IL-33 induces thymic involution-associated naive T cell aging and impairs host control of severe infection. Nat Commun 13(1): 6881.
- Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, et al. (2010) IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA 107(43): 18581.
- Yin H, Li X, Yuan B, Zhang B, Hu S, et al. (2011) Heme oxygenase-1 ameliorates LPS-induced acute lung injury correlated with downregulation of interleukin-33. International Immunopharmacology 11(12): 2112-2117.
- Ohno T, Oboki K, Morita H, Kajiwara N, Arae K, et al. (2011) Paracrine IL-33 stimulation enhances lipopolysaccharide-mediated macrophage activation. PLoS One 6(4): e18404.
- Xiang Y, Eyers F, Herbert C, Tay HL, Foster PS, et al. (2016) MicroRNA-487b Is a Negative Regulator of Macrophage Activation by Targeting IL-33 Production. Journal of Immunology 196(8): 3421.
- de Lima MHF, Hiroki CH, de Fátima Borges V, Cebinelli GCM, Santos J, et al. (2022) Sepsis-Induced Immunosuppression Is Marked by an Expansion of a Highly Suppressive Repertoire of FOXP3+ T-Regulatory Cells Expressing TIGIT. J Infect Dis 225(3): 531-541.
- Ke J, Cai G (2021) Effect of IL-33 on pyroptosis of macrophages in mice with sepsis via NF-κB/p38 MAPK signaling pathway. Acta Cir Bras 36(5): e360501.
- Ding X, Jin S, Shao Z, Xu L, Yu Z, et al. (2018) The IL-33-ST2 Pathway Contributes to Ventilator-Induced Lung Injury in Septic Mice in a Tidal Volume-Dependent Manner. Shock.
- Chang J, Xia Y, Wasserloos K, Deng M, Blose KJ, et al. (2017) Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS One 12(9): e0184770.
- Ding X, Tong Y, Jin S, Chen Z, Li T, et al. (2018) Mechanical ventilation enhances extrapulmonary sepsis-induced lung injury: role of WISP1–αvβ5 integrin pathway in TLR4-mediated inflammation and injury. Critical Care 22(1): 302.
- Li H-H, Li Q, Liu P, Liu Y, Li J, et al. (2012) WNT1-inducible signaling pathway protein 1 contributes to ventilator-induced lung injury. American Journal of Respiratory Cell and Molecular Biology 47(4): 528-535.
- Ding X, Wang X, Zhao X, Jin S, Tong Y, et al. (2015) RGD peptides protects against acute lung injury in septic mice through Wisp1-integrin β6 pathway inhibition. Shock 43(4): 352-360.
- Liu S, Deng M, Pan P, Turnquist HR, Pitt BR, et al. (2021) Mechanical Ventilation with Moderate Tidal Volume Exacerbates Extrapulmonary Sepsis-Induced Lung Injury via the IL33-WISP1 Signaling Pathway. Shock 56(3): 461-472.