Translational Perioperative and Pain Medicine (ISSN: 2330-4871)
ARTICLE DOI: 10.31480/2330-4871/056
REVIEW ARTICLE OPEN ACCESS
Neuron-glia interactions in pain
Jun-Ming Zhang
Pain Research Center, Department of Anesthesiology, University of Cincinnati College, of Medicine, 231 Albert Sabin Way, Cincinnati, OH 45267-0531, USA
Jun-Ming Zhang, MD, MSc, Pain Research Center, Department of Anesthesiology, University of Cincinnati College of Medicine. 231 Albert Sabin Way, Cincinnati, OH 45267-0531, Phone:+1-513-558-2427 Fax: +1-513-558-0995, E-mail: jun-ming.zhang@uc.edu
Editor: Yuan-Xiang Tao, PhD, MD, Professor and Vice Chair, Director, Center for Pain Medicine Research, Department of Anesthesiology. Rutgers, The State University of New Jersey, New Jersey Medical School, 185 S. Orange Ave., MSB, E-661 Newark, NJ 07103. Phone: 973-972-9812 (O) Fax: 973-972-1644. Email: yt211@njms.rutgers.edu
Received: June 01, 2017 | Accepted: June 13, 2017 | Published: June 17, 2017
Citation: Zhang JM. Neuron-glia interactions in pain. Transl Perioper & Pain Med 2017; 2(3): 14-22
Abstract
Glia contributes to pain by interacting with neighboring neurons in the spinal cord or sensory ganglion. There are two types of glia in the spinal cord: microglia and astrocyte, which are different from the satellite in sensory ganglion including dorsal root ganglion and trigeminal ganglion. Signals that mediate the two-way communication between glia cells and their adjacent neurons may include abnormal neuronal activity, calcium waves, cytokines, nitric oxide, and growth factors. These highly localized paracrine signals may account for the observation that both injured and non-injured neurons in the same ganglion play important roles in pain behaviors in partial nerve injury models of chronic pain. In this review, we will describe roles of spinal glia and satellite glia in pain resulting from peripheral nerve injury or local inflammation followed by discussion of the mechanisms of neuron-glia communications in pain. Relatively more attention is focused on the neuron-glia interactions in the sensory ganglion.
Keywords
Neuron, Glia, Pain, Mechanism
Spinal glial cells play key roles in pathological pain
Recent studies indicate that activated glial cells in spinal cord and dorsal root ganglion (DRG) play key roles in chronic pain conditions [1-5]. There are two main types of glial cells in the central nervous system (CNS), microglia and astrocytes. Long viewed as a mere matrix for neurons, they have recently been found to play complex and important roles in regulation of the local neuronal environment and the synapse. In response to neuronal damage or inflammatory stimulation, the glial cells become activated, undergo gliosis, and release a number of signals. This communication between spinal/sensory neurons and glia, which may be highly localized or paracrine, can occur through a number of messengers. These include excitatory amino acid transmitters [6], substance P (SP) [7], prostaglandins [8], adenosine triphosphate (ATP) [9], nitric oxide (NO) [10], and fractalkine [11,12]. Activated glia are known to release a number of key pro-inflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin (IL)-1β, and IL-6 [13-16].
It has been well documented that glial activation in the spinal cord is critical for the initiation and persistence of neuropathic pain [17-24]. Spinal administration of fractalkine produces cutaneous mechanical hyperalge-sia, whereas blocking spinal fractalkine receptor using selective antagonist alleviates neuropathic pain [25]. Furthermore, neurogenic pain induced by topical ap-plication of zymosan can be blocked by a glial inhibi-tor fluorocitrate [18,26-28]. Similarly, administration of another glial inhibitor, minocycline [29-31], also blocks neuropathic pain [32,33].
Following peripheral nerve injury, the two types of glial cells in the spinal cord also undergo morpho-logical changes. Microglial cells proliferate, while astrocytes change in shape and manifest increased expression of GFAP [25,34]. Blocking neuronal activity originating from the injured DRG neurons suppressed microglia activation in the spinal cord [35]. It suggests that peripheral activity may contrib-ute to the activation of spinal glia.
Roles of satellite glia in the DRG in pathological pain
The satellite glial cells (SGC) in the sensory ganglia are different from those in the central nervous system (CNS). Though they share many characteristics, these SGC do not fit neatly into the astrocyte/microglia classification scheme, showing some ultrastructural characteristics of both. DRG neurons lack dendrites, and each neuron is wrapped with its own sheath of SGC and connective tissue, a structure unique to the DRG [26,37].
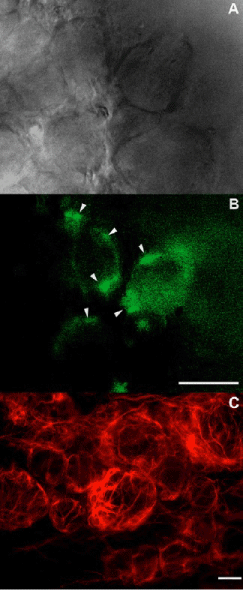
Changes in SGC in pathological pain models have been well-described, though not as extensively as in the CNS [38]. Following peripheral nerve injury, the SGC that surround the soma of the axotomized sen-sory neurons proliferate [39-41], elaborate processes [42], and become immunoreactive (IR) for GFAP [43]. The expression of GFAP is different between glia in DRG and CNS; in the CNS, some GFAP expression is observed even under normal conditions, while in DRG, the expression of GFAP is not detected until some form of nerve injury occurs (Figure 1). SGC activation is also observed in DRGs with localized inflammation without peripheral nerve injury [44] and in DRGs with chronic compression [45].
Figure 1: TActivated satellite glia in axotomized DRGs. A & B: Visualizing reactive satellite glial cells surrounding live DRG neurons in an in vitro nerve-DRG preparation in mouse. Satellite glia are activated by sciatic nerve section 2 weeks prior. A: The surface of a DRG visualized under an up-right light microscope. B: Same DRG visualized under a fluorescent microscope. Scale bar=50 μm. Arrows indicate activated SGC. C: GFAP immunoreactivity in a rat DRG section showing GFAP-positive SGCs surrounding nerve-injured DRG neurons. Scale bar=20 μm.
Satellite glial activation in paravertebral sympathetic ganglia
While much works on glia have been focused on the spinal cord and the DRG, there has been little attention about glial activation in the sympathetic ganglia. Since post-sympathetic efferents travel with peripheral nerves, it is anticipated that injury to the peripheral nerve also damages axons of the sympathetic nerves in addition to the sensory and motor fibers. One early study compared the effects of sciatic nerve transection on neuroinflammatory responses in DRG as compared to lumbar sympathetic ganglia. They report that GFAP level, macrophage immunoreactivity, and T cell re-sponses were even stronger in the sympathetic ganglia than in the DRGs [46].
Our lab recently investigated changes in T cells, mac-rophage responses and GFAP expression in lumbar sympathetic ganglia in the SNL model [47]. In this model, the injured L5 spinal nerve contains post-sym-pathetic axons. To examine inflammatory responses in the sympathetic ganglia with remote inflammation without nerve injury, we also compared the effects of spinal nerve ligation (SNL) on these neuroinflamma-tory markers to the effects observed in the radicular pain model after local inflammation of the L5 DRG. Neuroinflammation in the inflamed DRG is strong-ly mitigated by cutting the gray rami, the source of sympathetic fibers in the spinal nerve and nearby DRG [48]. It was found that compared to local DRG in-DRG.
The roles of satellite glia activation in the parasym-pathetic ganglia are not clear. It is also unknown whether neuron and glia interact in a similar way as they do in the sensory ganglion. It is anticipated that satellite glial activation may contribute to enhanced excitability of the sympathetic neurons after sensory nerve injury or localized DRG inflammation as recent-ly demonstrated by our group [48]. Since sympathetic activity is reported to regulate immune homeostasis under physiological conditions [49], altered sympa-thetic neuron properties may, in turn, contribute to inflammatory responses in the sensory ganglion after peripheral nerve injury or local DRG inflammation.
Roles of satellite glia in communication between injured and non-injured neurons in partial nerve injury
The sciatic nerve, which arises from neurons primarily in the L4/L5 DRG, is commonly used for partial nerve injury models such as the Seltzer [50] and CCI [51] models. These partial peripheral nerve injury mod-els were developed in order to model certain clinical situations more closely than nerve transection models. Studies using these models have shown the impor-tance of uninjured neurons for maintained pathological pain behaviors [52], and have shown that uninjured neurons as well as injured neurons undergo changes in electrical properties and gene or protein expression [52-55]. This raises the question of how informa-tion about the injury is "transmitted" to the uninjured cells. Some possible mechanisms include electrical cross-excitation between neurons [56] and exposure of intact axons to Wallerian degeneration in adjacent injured axons (for review [57]). However, it has been proposed that some of the SGC mechanisms may also provide a pathway for injured neurons to communi-cate with and alter the properties of uninjured neurons [43,58,59]. For example, in the trigeminal gangli-on, where sensory neurons have a more somatotopic organization than in the DRG, it was observed that injury to a particular tooth caused glia activation not only around the damaged neurons innervating that tooth (as demonstrated by both anatomical location and retrograde tracers), but also around some adjacent uninjured neurons [59].
Possible mechanisms of neuronglia communications
Evidence from single recordings of the SGC and neurons suggests that neuronal activity and glial cell activation are intricately linked [60-62]. A number of plausible mechanisms underlying neuron-glia commu-nications has been suggested. First, K+ accumulation in the extracellular space between neurons and the sheath formed by surrounding SGC [5]. In cultured spinal astrocytes, elevation of [K+] results in altered GFAP expression [63]. Depolarization or hyperpo-larization of glial cell membranes has been observed in response to changes in adjacent neurons [64]. On the other hand, satellite glial cells may affect neuro-nal excitability/activity through similar mechanisms [65,66]. These membrane responses in glial cells may be associated with the concentration of ions in the glia. Second, spontaneously active neurons may also influence adjacent SGC by local release of SP, ATP, or other messengers [67-72]. Third, in response to gluta-mate application or electrical stimulation, glia respond with slow alternating flows of calcium ions into and out of the cells, which has been described as "calcium waves" [73,74]. Thus, calcium wave is believed to be another method for glial cells to communicate with nearby neurons. Lastly, abnormal spontaneous ac-tivity of the injured or inflamed sensory neurons may affects surrounding SGC by regulating the synthesis release of neurotrophins.
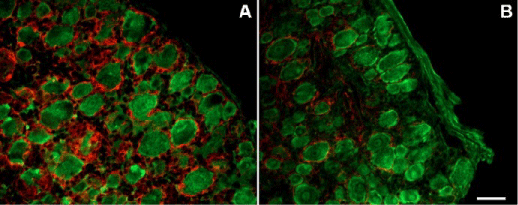
As discussed above, abnormal electrical activity may be a key way for injured neurons to communicate with their surrounding glia. In commonly used animal models of neuropathic pain, abnormal spontaneous ac-tivity of neurons and pain behaviors both appear with-in the first 12 hours to 2 days post injury [50,59,75]. Most other pathological changes described above, including glial activation, begin later than this. Spon-taneous afferent activity is therefore a likely candidate for initiating chronic pain. A key observation is that temporarily blocking spontaneous activity beginning at the time of injury reduces or eliminates spontaneous pain, hyperalgesia, and allodynia. This has been demonstrated in several different pain models, using methods to suppress spontaneous activity that vary widely in their specific targets [76-80]. Data from our own group showed that, in two different partial nerve injury models, early temporary blockade of sponta-neous activity was sufficient to permanently prevent development of pain behaviors [81]. In another study [35], we tested effects of suppressing DRG neuron ac-tivity with sodium channel blockers on SGC activation in the rat SNL model, and found that local perfusion of the axotomized DRG with tetrodotoxin significant-ly reduced this activation as evidenced by decreased GFAP immunoreactivity (Figure 2). Similar findings were made with a more distal nerve injury (the spared nerve injury or SNI model), using a local anesthetic (the depot form of bupivacaine) at the nerve injury site. Local perfusion of the DRG in the SNL model also significantly reduced microglia (OX-42 immuno-reactivity) and astrocytic (GFAP immunoreactivity) activations in the adjacent spinal cord. Results from these experiments indicate that early abnormal sponta-neous activity from injured sensory neurons may play important roles in glial activation as well as patholog-ical pain.
Figure 2: Neuronal blockade with TTX reduced satellite glia activation after spinal nerve ligation. Sections of DRG stained for GFAP (red) and Neu-N (Green). A: GRAP-immunoreactive satellite glia activation following SNL on POD3; B: Applying TTX locally to the axotomized DRG starting at the time of nerve injury reduced the nerve injury-induced GFAP expression. Scale bar = 50 μm. Modified from Xie et al., Neuroscience 2009 (35).
Messengers for neuron-glia communication
A number of different messengers may serve for the communication between sensory neurons and sur-rounding/adjacent glia. These include:
Cytokines/chemokines: At the level of the DRG, a number of different cytokines and their receptors have been shown to be increased in response to various experimental pathologic conditions [82-85]. In a few cases, the SGC have been shown to be the source of particular cytokines or receptors (e.g., TNF-α [36]), but in most cases the cell of origin is not well defined. Fractalkine also known as chemokine (C-X3-C motif) ligand 1 is a protein that in humans is encoded by the CX3CL1 gene. Unlike the spinal cord, in the DRG, fractalkine is constitutively expressed by the primary nociceptive neurons. Increased SGC activation as indi-cated by increased GRAP mRNA expression following carrageenan injection in the hind paw was inhibited by anti-fractalkine antibody administered into the DRG [86]. These findings suggest that fractalkine released by nociceptive neurons contributes to the genesis of inflammatory pain through the activation of the SGCs. SGC are also responsive to other inflammatory media-tors such as bradykinin and ATP. Both SGC and DRG neurons can synthesize and respond to prostaglandins, another inflammatory mediator, though much of the evidence is from cultured neurons and SGC [36].
NO/cGMP: The nitric oxide signaling system is one of the best defined in DRG. Some neurons express NO synthase, and many SGC contain cGMP cyclase, the enzyme activated by NO. NO is released in a non-vesicular fashion and can diffuse short distances between cells. The SGC also accumulate NO precur-sors [87], and this system has been proposed as a like-ly candidate for neuron-SGC signaling. Components of the system are upregulated after neuronal injury, and some but not all investigators report reduction of pain behaviors by NO inhibitors (for review see [36]).
Neurotrophins: Neurotrophins such as nerve growth factor (NGF), neurotrophin (NT)-3, and brain-derived neurotrophic factor (BDNF) may play roles in chronic pain states in addition to their roles in neuronal devel-opment and survival. SGC express NGF receptors and may express NGF and NT-3, especially after injury. These have been proposed to play a role in injury-in-duced sympathetic sprouting in the DRG [88-90]. Hence these molecules represent another possible communication route between neurons and SGC.
Glial coupling
Several studies have demonstrated that nerve injury leads to increased coupling (indicated by diffusion of injected dye) among SGC around an individual neuron, and among SGC surrounding neighboring neurons [91,92]. The functional significance of this coupling, and its relationship to glial activation, are not yet clear. However, these observations may reflect another mechanism by which glia mediate abnormal communication between neurons following nerve injury and may contribute to cross-talk among DRG neurons after nerve injury [93].
Summary
Glial cells in the spinal cord and sensory ganglion react to peripheral nerve injury or inflammation, and contribute to pain by interacting with neighboring neurons. Preclinical studies have demonstrated ro-bust effects of inhibiting glial activation in alleviating pain. However, clinical trials using glial inhibitors for managing neuropathic pain have been generally disappointing. Much more works remain to be done to further understand neuro-glia interactions in pain and to translate preclinical studies to clinical pain manage-ment.
Acknowledgements
None
Disclosure of Funding
Supported in part by National Institutes of Health grants AR0068989, NS045594 and NS055860.
Conflict Interests Disclosure
The authors have no conflicting interests to disclose.
References
- Huang LY, Gu Y, Chen Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013;61:1571-81.
- Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain 2013;154 Suppl 1:S10-28.
- Mika J, Zychowska M, Popiolek-Barczyk K et al.Importance of glial activation in neuropathic pain. Eur J Pharmacol 2013;716:106-19.
- Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol 2008;21:570-9.
- Ren K, Dubner R. Interactions between the im-mune and nervous systems in pain. Nat Med 2010;16:1267-76.
- Takuma K, Matsuda T, Hashimoto H et al. Role of Na(+)-Ca2+ exchanger in agonist-induced Ca2+ signaling in cultured rat astrocytes. J Neurochem 1996;67:1840-5.
- Marriott DR, Wilkin GP, Wood JN. Substance P-in-duced release of prostaglandins from astrocytes: re-gional specialisation and correlation with phospho-inositol metabolism. J Neurochem 1991;56:259-65.
- Bezzi P, Carmignoto G, Pasti L et al. Prostaglan-dins stimulate calcium-dependent glutamate release in astrocytes. Nature 1998;391:281-5.
- Hide I, Tanaka M, Inoue A et al. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 2000;75:965-72
- Mollace V, Colasanti M, Muscoli C et al. The effect of nitric oxide on cytokine-induced release of PGE2 by human cultured astroglial cells. Br J Pharmacol 1998;124:742-6.
- Chapman GA, Moores K, Harrison D et al. Fractal-kine cleavage from neuronal membranes represents an acute event in the inflammatory response to ex-citotoxic brain damage. J Neurosci 2000;20:RC87.
- Maciejewski-Lenoir D, Chen S, Feng L et al.Characterization of fractalkine in rat brain cells:migratory and activation signals for CX3CR-1-ex-pressing microglia. J Immunol 1999;163:1628-35.
- Winkelstein BA, Rutkowski MD, Sweitzer SM et al. Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. J CompNeurol 2001;439:127-39.
- Kyrkanides S, Olschowka JA, Williams JP et al.TNF alpha and IL-1beta mediate intercellular adhe-sion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J Neuroimmu-nol 1999;95:95-106.
- Chao CC, Hu S, Peterson PK. Glia, cytokines, and neurotoxicity. Crit Rev Neurobiol 1995;9:189-205.
- Watkins LR, Maier SF. Beyond neurons: Evidence that immune and glial cells contribute to pathologi-cal pain states [Review]. Physiol Rev 2002;82:981-1011.
- DeLeo JA, Rutkowski MD, Stalder AK, Campbell IL. Transgenic expression of TNF by astrocytes increases mechanical allodynia in a mouse neurop-athy model. Neuroreport 2000;11:599-602.
- Milligan ED, Twining C, Chacur M et al. Spi-nal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 2003;23:1026-40.
- Sweitzer SM, Colburn RW, Rutkowski M, DeLeo JA. Acute peripheral inflammation induces moder-ate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res 1999;829:209-21.
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial acti-vation and neuropathic pain behavior. Exp Neurol 1999;157:289-304.
- Colburn RW, DeLeo JA, Rickman AJ et al. Dis-sociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol 1997;79:163-75.
- Rutkowski MD, Winkelstein BA, Hickey WF et al. Lumbar nerve root injury induces central nervous system neuroimmune activation and neuroinflam-mation in the rat: relationship to painful radiculop-athy. Spine 2002;27:1604-13.
- Tsuda M, Beggs S, Salter MW, Inoue K. Microglia and intractable chronic pain. Glia 2013;61:55-61.
- Old EA, Clark AK, Malcangio M. The role of glia in the spinal cord in neuropathic and inflammatory pain. Handb Exp Pharmacol 2015;227:145-70.
- Watkins LR, Milligan ED, Maier SF. Glial proin-flammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol 2003;521:1-21.
- Meller ST, Dykstra C, Grzybycki D et al. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuro-pharmacology 1994;33:1471-8.
- Milligan ED, O'Connor KA, Nguyen KT et al. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spi-nal cord proinflammatory cytokines. J Neurosci 2001;21:2808-19.
- Milligan ED, Mehmert KK, Hinde JL et al. Ther-mal hyperalgesia and mechanical allodynia pro-duced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glyco-protein, gp120. Brain Res 2000;861:105-16.
- Wu DC, Jackson-Lewis V, Vila M et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosc 2002;22:1763-71.
- Yrjanheikki J, Keinanen R, Pellikka M et al.Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. PNAS 1998;95:15769-74.
- Yrjanheikki J, Tikka T, Keinanen R et al. A tet-racycline derivative, minocycline, reduces in-flammation and protects against focal cerebral ischemia with a wide therapeutic window. PNAS 1999;96:13496-500.
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 2001;166:7527-33.
- Lin S, Zhang Y, Dodel R et al. Minocycline blocks nitric oxide-induced neurotoxicity by inhibition p38 MAP kinase in rat cerebellar granule neurons. Neurosci Lett 2001;315:61-4.
- Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain. Neurochem Int 2004;45:389-95.
- Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain. Neurochem Int 2004;45:389-95.
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev 2005;48:457-76.
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev 2005;48:457-76.
- Hanani M. Role of satellite glial cells in gastroin-testinal pain. Front Cell Neurosci 2015;9:412.
- Barron KD, Marciano FF, Amundson R, Mankes R. Perineuronal glial responses after axotomy of central and peripheral axons. A comparison. Brain Res 1990;523:219-29.
- Gehrmann J, Monaco S, Kreutzberg GW. Spinal cord microglial cells and DRG satellite cells rap-idly respond to transection of the rat sciatic nerve. Restor Neurol Neurosci 1991;2:181-98.
- Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci 1991;11:972-8.
- Leech RW. Changes in satellite cells of rat dorsal root ganglia during central chromatolysis. An elec-tron microscopic study. Neurology 1967;17:349-58.
- Woodham P, Anderson PN, Nadim W, Turmaine M. Satellite cells surrounding axotomised rat dorsal root ganglion cells increase expression of a GFAP-like protein. Neurosci Lett 1989;98:8-12.
- Xie WR, Deng H, Li H et al. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflam-matory irritation of the spinal ganglia. Neurosci-ence 2006;142:809-22.
- Zhang H, Mei X, Zhang P et al. Altered function-al properties of satellite glial cells in compressed spinal ganglia. Glia 2009;57:1588-99.
- Hu P, McLachlan EM. Inflammation in sympathetic ganglia proximal to sciatic nerve transection in rats. Neurosci Lett 2004;365:39-42.
- Li JY, Xie W, Strong JA et al. Mechanical hy-persensitivity, sympathetic sprouting, and glial activation are attenuated by local injection of corticosteroid near the lumbar ganglion in a rat model of neuropathic pain. Reg Anesth Pain Med 2011;36:56-62.
- Li AL, Zhang JD, Xie W et al. Inflammatory Changes in Paravertebral Sympathetic Ganglia in Two Rat Pain Models. Neurosci Bull 2017.
- Xie W, Chen S, Strong JA et al. Localized sympa-thectomy reduces mechanical hypersensitivity by restoring normal immune homeostasis in rat mod-els of inflammatory pain. J Neurosci 2016;36:8712-25.
- Seltzer Z, Dubner R, Shir Y. A novel behav-ioral model of neuropathic pain disorders pro-duced in rats by partial sciatic nerve injury. Pain 1990;43:205-18.
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988;33:87-107
- Obata K, Yamanaka H, Fukuoka T et al. Contribu-tion of injured and uninjured dorsal root ganglion neurons to pain behavior and the changes in gene expression following chronic constriction injury of the sciatic nerve in rats. Pain 2003;101:65-77.
- Tsuzuki K, Kondo E, Fukuoka T et al. Differential regulation of P2X(3) mRNA expression by periph-eral nerve injury in intact and injured neurons in the rat sensory ganglia. Pain 2001;91:351-60.
- Michaelis M, Liu X, Janig W. Axotomized and intact muscle afferents but no skin afferents de-velop ongoing discharges of dorsal root ganglion origin after peripheral nerve lesion. J Neurosci 2000;20:2742-8.
- Ma W, Bisby MA. Increase of preprotachykinin mRNA and substance P immunoreactivity in spared dorsal root ganglion neurons following partial sci-atic nerve injury. Eur J Neurosci 998;10:2388-99.
- Liu CN, Amir R, Devor M. Effect of age and nerve injury on cross-excitation among sensory neurons in rat dorsal root ganglia. Neurosci Lett 1999;259:95-8.
- Campbell JN. Nerve lesions and the generation of pain. Muscle & Nerve 2001;24:1261-73.
- Fenzi F, Benedetti MD, Moretto G, Rizzuto N. Glial cell and macrophage reactions in rat spinal ganglion after peripheral nerve lesions: an immu-nocytochemical and morphometric study. Arch Ital Biol 2001;139:357-65.
- Stephenson JL, Byers MR. GFAP immunoreactiv-ity in trigeminal ganglion satellite cells after tooth injury in rats. Exp Neurol 1995;131:11-22.
- Schmidt J, Prinz P, Deitmer JW. Glial hyperpolar-ization upon nerve root stimulation in the leech Hirudo medicinalis. GLIA 1999;27:32-8.
- Gunzel D, Schlue WR. Mechanisms of Mg2+ influx, efflux and intracellular 'muffling' in leech neurones and glial cells. Magnesium Res 2000;13:123-38.
- Murphy TH, Blatter LA, Wier WG, Baraban JM. Rapid communication between neurons and astrocytes in primary cortical cultures. J Neurosci 1993;13:2672-9.
- Canady KS, Ali-Osman F, Rubel EW. Extracellular potassium influences DNA and protein syntheses and glial fibrillary acidic protein expression in cultured glial cells. Glia 1990;3:368-74.
- Lohr C, Deitmer JW. Dendritic calcium tran-sients in the leech giant glial cell in situ. GLIA 1999;26:109-18.
- Lohr C, Deitmer JW. Dendritic calcium tran-sients in the leech giant glial cell in situ. GLIA 1999;26:109-18.
- Rouach N, Glowinski J, Giaume C. Activity-depen-dent neuronal control of gap-junctional communi-cation in astrocytes. J Cell Biol 2000;149:1513-26.
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci 2006;7:423-36.
- Sanada M, Yasuda H, Omatsu-Kanbe M et al.Increase in intracellular Ca(2+) and calcitonin gene-related peptide release through metabotropic P2Y receptors in rat dorsal root ganglion neurons. Neuroscience 2002;111:413-22.
- Nakatsuka T, Mena N, Ling J, Gu JG. Depletion of substance P from rat primary sensory neurons by ATP, an implication of P2X receptor-mediated re-lease of substance P. Neuroscience 2001;107:293-300.
- Rydh-Rinder M, Kerekes N, Svensson M, Hokfelt T. Glutamate release from adult primary sensory neurons in culture is modulated by growth factors. Regul Pept 2001;102:69-79.
- Fields RD, Stevens B. ATP: an extracellular sig-naling molecule between neurons and glia. Trends Neurosci 2000;23:625-33.
- Skoff AM, Resta C, Swamydas M, Adler JE. Nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF) regulate substance P release in adult spinal sensory neurons. Neurochem Res 2003;28:847-54.
- Venance L, Piomelli D, Glowinski J, Giaume C. Inhibition by anandamide of gap junctions and in-tercellular calcium signalling in striatal astrocytes. Nature 1995;376:590-4.
- Dani JW, Chernjavsky A, Smith SJ. Neuronal activ-ity triggers calcium waves in hippocampal astro-cyte networks. Neuron 1992;8:429-40.
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spi-nal nerve ligation in the rat. Pain 1992;50:355-63
- Lai J, Gold MS, Kim CS et al. Inhibition of neuro-pathic pain by decreased expression of the tetro-dotoxin-resistant sodium channel, Na(v)1.8. Pain 2002;95:143-52.
- Yoon YW, Na HS, Chung JM. Contributions of injured and intact afferents to neuropathic pain in an experimental rat model. Pain 1996;64:27-36.
- Lyu YS, Park SK, Chung K, Chung JM. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res 2000;871:98-103.
- Chaplan SR, Guo HQ, Lee DH et al. Neuronal hy-perpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci 2003;23:1169-78.
- Xiao WH, Bennett GJ. Synthetic omega-conopep-tides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther 1995;274:666-72.
- Xie W, Strong JA, Meij JT et al. Neuropathic pain: Early spontaneous afferent activity is the trigger. Pain 2005;116:243-56.
- Osamura N, Ikeda K, Ito T et al. Induction of interleukin-6 in dorsal root ganglion neurons after gradual elongation of rat sciatic nerve. Exp Neurol 2005;195:61-70.
- Li Y, Ji A, Weihe E, Schafer MK. Cell-specific expression and lipopolysaccharide-induced regula-tion of tumor necrosis factor alpha (TNFalpha) and TNF receptors in rat dorsal root ganglion. J Neuro-sci 2004;24:9623-31.
- Murata Y, Onda A, Rydevik B et al. Distribution and appearance of tumor necrosis factor-alpha in the dorsal root ganglion exposed to experimental disc herniation in rats. Spine 2004;29:2235-41.
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upreg-ulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 2004;29:1082-8.
- Souza GR, Talbot J, Lotufo CM et al. Fractalkine mediates inflammatory pain through activation of satellite glial cells. Proc Natl Acad Sci U S A 2013;110:11193-8.
- Aoki E, Takeuchi IK, Shoji R, Semba R. Localiza-tion of nitric oxide-related substances in the periph-eral nervous tissues. Brain Res 1993;620:142-5.
- Zhou XF, Deng YS, Chie E et al. Satellite-cell-de-rived nerve growth factor and neurotrophin-3 are involved in noradrenergic sprouting in the dorsal root ganglia following peripheral nerve injury in the rat. Eur J Neurosci 1999;11:1711-22.
- Walsh GS, Krol KM, Kawaja MD. Absence of the p75 neurotrophin receptor alters the pattern of sym-pathosensory sprouting in the trigeminal ganglia of mice overexpressing nerve growth factor. J Neuros-ci 1999;19:258-73.
- Ramer MS, Thompson SW, McMahon SB. Causes and consequences of sympathetic basket formation in dorsal root ganglia. Pain 1999;Suppl 6:S111-20.
- Huang TY, Cherkas PS, Rosenthal DW, Hanani M.Dye coupling among satellite glial cells in mamma-lian dorsal root ganglia. Brain Res 2005;1036:42-9.
- Hanani M, Huang TY, Cherkas PS et al. Glial cell plasticity in sensory ganglia induced by nerve dam-age. Neuroscience 2002;114:279-83.
- Kim YS, Anderson M, Park K et al. Coupled Acti-vation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 2016;91:1085-96.
Table of Contents
- Abstract
- Keywords
- Spinal glial cells play key roles in pathological pain
- Roles of satellite glia in the DRG in pathological pain
- Satellite glial activation in paravertebral sympathetic ganglias
- Roles of satellite glia in communication between injured and non-injured neurons in partial nerve injury
- Possible mechanisms of neuronglia communications
- Messengers for neuron-glia communication
- Glial coupling
- Summary
- Acknowledgements
- Disclosure of Funding
- Conflict Interests Disclosure
- Figure 1
- Figure 2
- References